
 Descargar número completo
Descargar número completo Download full issue
Download full issueCITA ESTE TRABAJO
López Ortega S, Parra López B, Alonso Belmonte C, González Grande R, Jiménez Pérez M. Enfermedad Gastrointestinal Relacionada Con La IgG4. RAPD 2026;49(2):54-63. DOI: 10.37352/2026492.2
Introducción
La enfermedad relacionada con IgG4 (IgG4-RD) fue reconocida como entidad propia a comienzos de este siglo tras la descripción de concentraciones séricas elevadas de IgG4 en pacientes con pancreatitis esclerosante[1]. En la actualidad se considera una enfermedad fibroinflamatoria sistémica capaz de afectar a numerosos órganos, entre ellos páncreas, vía biliar, hígado, glándulas salivales, riñones, retroperitoneo, pulmones y tracto gastrointestinal[2],[3],[8]. Su relevancia clínica radica en su capacidad para simular neoplasias u otras enfermedades inflamatorias y en el riesgo de fibrosis irreversible cuando el diagnóstico y tratamiento se retrasan[2],[8],[27].
Desde el punto de vista digestivo, la pancreatitis autoinmune tipo 1 constituye la manifestación más representativa, pero no la única. La colangitis esclerosante asociada a IgG4, la afectación hepática, la infiltración del tubo digestivo y la mesenteritis esclerosante amplían el espectro clínico y plantean retos diagnósticos relevantes para el gastroenterólogo[4]-[7],[12].
El objetivo de esta revisión es actualizar los aspectos epidemiológicos, patogénicos, diagnósticos y terapéuticos de la IgG4-RD, con énfasis en las manifestaciones gastrointestinales y en los elementos prácticos de mayor utilidad clínica.
Enfermedad relacionada con IgG4
La IgG4-RD es una enfermedad sistémica inmunomediada caracterizada por infiltración linfoplasmocítica rica en células plasmáticas IgG4+, fibrosis y, según el órgano, flebitis obliterativa[2],[8],[11]. Su presentación es heterogénea y depende de la localización anatómica y del número de órganos afectados. Más de la mitad de los pacientes presentan compromiso multiorgánico, y la evolución prolongada puede conllevar daño estructural irreversible[8],[12],[27].
Epidemiología
La enfermedad aparece predominantemente en adultos entre la quinta y séptima décadas de la vida, aunque se han descrito casos pediátricos y en edades avanzadas[5],[7]. Existe predominio masculino en las formas pancreatobiliares y retroperitoneales, mientras que la afectación de cabeza y cuello puede ser relativamente más frecuente en mujeres[5],[8]. La incidencia y prevalencia probablemente están infradiagnosticadas. Estudios poblacionales recientes en Estados Unidos estiman una incidencia aproximada de 0,78-1,39 casos por 100.000 personas-año y una prevalencia creciente, en paralelo con el mayor reconocimiento de la enfermedad[9].
Patogenia
La patogenia de la IgG4-RD es compleja y combina susceptibilidad genética, exposición antigénica repetida y una respuesta inmune desregulada[2],[8],[10]. Entre los mecanismos implicados destacan la activación de linfocitos T helper foliculares, que favorecen el cambio de isotipo en células B hacia IgG4 e IgE, y la interacción entre células B, plasmablastos y subpoblaciones T efectoras en tejidos diana[8],[10].
Estudios recientes han consolidado el papel central de las células T CD4+ citotóxicas clonales, en particular aquellas con fenotipo SLAMF7+, como principales efectoras del daño tisular[10],[27]. Estas células producen citocinas profibróticas como TGF-β e IFN-γ, presentan expansión clonal en sangre periférica y tejidos afectados y se correlacionan con la actividad de la enfermedad[10],[27]. Se ha planteado además que parte del efecto terapéutico de rituximab podría depender de la interrupción del soporte antigénico que las células B proporcionan a estas poblaciones T efectoras[2],[10],[27].
Asimismo, los plasmablastos circulantes han emergido como un biomarcador dinámico de actividad inflamatoria. Su cuantificación parece ofrecer mayor sensibilidad que la IgG4 sérica en algunos contextos, incluso en pacientes con niveles normales de esta inmunoglobulina, lo que refuerza su potencial utilidad para monitorización[10],[26],[27].
Pese a la asociación entre la enfermedad y la elevación de IgG4 sérica y tisular, el papel exacto de la IgG4 en la patogenia sigue siendo incierto. En lugar de ser necesariamente la mediadora directa del daño, podría actuar como marcador de una respuesta inmune anómala más compleja[8],[10].
Histopatología
La histopatología constituye un pilar del diagnóstico. Las tres características clásicas son: infiltrado linfoplasmocítico denso, fibrosis estoriforme y flebitis obliterativa[8],[11]. La expresión de cada una de ellas varía según el órgano afectado, y no siempre están presentes simultáneamente. El infiltrado linfoplasmocítico difuso representa uno de los hallazgos más característicos en el estudio histológico (Figura 1), mientras que la fibrosis estoriforme constituye otro de los rasgos morfológicos más representativos de la entidad (Figura 2)[8],[11]. La flebitis obliterativa, cuando está presente, aporta además un gran peso diagnóstico (Figura 3)[8],[11]. La inmunotinción demuestra incremento de células plasmáticas IgG4+ y una relación IgG4+/IgG+ elevada, habitualmente superior al 40%, si bien el punto de corte debe interpretarse de acuerdo con el tejido analizado[8],[11].
Figura 1
Imagen histológica en la se observa un denso y difuso infiltrado linfoplasmocítico. Cortesía de la Dra Teresa Pereda, Hospital Costa del Sol (Marbella).
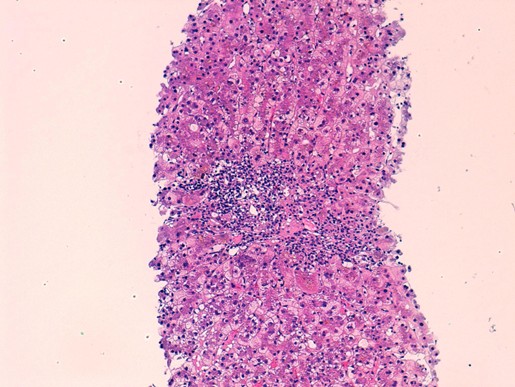
Figura 2
Imagen histológica en la que se objetiva tejido fibrótico arremolinado que envuelve a un infiltrado celular en fibrillas de colágeno (A). En la imagen B se observa fibrosis estoriforme rodeada de un infiltrado linfoplasmocítico.
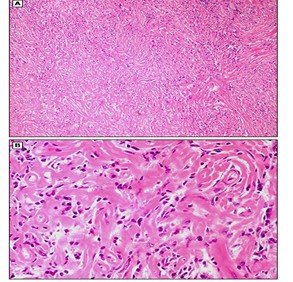
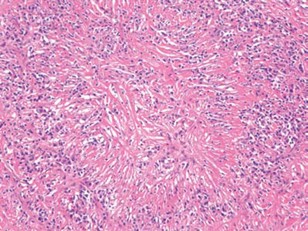
Figura 3
Imagen histológica en la que se muestra obliteración parcial o completa de las venas medianas, secundaria a la infiltración linfoplasmocítica, compatible con flebitis obliterativa.
La fibrosis estoriforme representa una disposición arremolinada de haces de colágeno, especialmente útil para el diagnóstico en órganos profundos[11]. Debe tenerse en cuenta que la distribución parcheada de las lesiones puede limitar el rendimiento de las biopsias pequeñas, especialmente en muestras obtenidas con aguja[11],[15].
Serología y biomarcadores
La mayoría de los pacientes presentan elevación sérica de IgG4, aunque esta puede ser normal en un porcentaje no despreciable de casos[5],[8],[26]. Además, el aumento de IgG4 sérica no es específico y puede observarse en otras enfermedades inflamatorias, infecciosas o neoplásicas[5],[8],[26]. Por ello, su utilidad es complementaria y nunca suficiente para confirmar el diagnóstico de forma aislada.
Además de la IgG4 sérica, se han descrito otras alteraciones inmunológicas de interés. La eosinofilia periférica y el aumento de IgE sérica son hallazgos relativamente frecuentes y reflejan un perfil inmunológico predominantemente Th2[10],[26]. En pacientes con afectación renal puede observarse hipocomplementemia, con descenso de C3 y C4, asociada a mayor actividad inflamatoria[26]. No obstante, en la actualidad no existe un biomarcador único con sensibilidad y especificidad suficientes para establecer por sí solo el diagnóstico de IgG4-RD[2],[10],[26].
Presentación clínica
La expresión clínica de la enfermedad es muy variable y, con frecuencia, insidiosa[2],[5],[8]. Entre las manifestaciones generales destacan astenia, pérdida de peso, febrícula, dolor o aumento de tamaño del órgano afectado y, en algunos casos, síndrome constitucional[5],[8],[12]. Este perfil inespecífico obliga a considerar un amplio diagnóstico diferencial, que incluye neoplasias, linfomas, infecciones y otras enfermedades autoinmunes o fibroinflamatorias[2],[8][27].
Pruebas de imagen y criterios de clasificación
Las técnicas de imagen desempeñan un papel fundamental tanto en el diagnóstico como en la evaluación de la extensión y en el seguimiento[4][5],[13],[16]. La tomografía computarizada (TC) con contraste resulta útil en la valoración inicial, especialmente en páncreas, vía biliar, retroperitoneo y grandes vasos [4],[5],[16]. La resonancia magnética y la colangiorresonancia (Colangio-RM) aportan una mejor caracterización tisular y ductal[4],[5],[16]. La tomografía por emisión de positrones (PET-TC) puede identificar afectación multiorgánica subclínica y es especialmente valiosa en enfermedad sistémica[2],[4]. La ecoendoscopia permite obtener muestras tisulares dirigidas en páncreas y vía biliar [4],[13].
En 2019, el American College of Rheumatology y la European League Against Rheumatism desarrollaron criterios de clasificación con el objetivo de homogeneizar la inclusión de pacientes en estudios clínicos[3]. Estos criterios fueron diseñados con fines clasificatorios y no estrictamente diagnósticos, por lo que su aplicación en la práctica clínica debe contextualizarse dentro de una evaluación integral[2],[3]. El diagnóstico de IgG4-RD continúa sustentándose en la combinación de hallazgos clínicos, serológicos, radiológicos e histopatológicos, con exclusión de entidades mimetizadoras[2],[3],[8]).
Diagnóstico general
En el contexto de la afectación pancreática, los criterios HISORt mantienen una gran utilidad clínica, especialmente cuando la confirmación histológica no es factible[13],[15]. Este enfoque integra histología, imagen, serología, afectación de otros órganos y respuesta al tratamiento[13],[15]. La respuesta a glucocorticoides, en ausencia de datos sugestivos de malignidad, sigue siendo un elemento orientador relevante, aunque nunca debe utilizarse de forma aislada si persiste la sospecha de cáncer[2],[13],[15].
Manifestaciones gastrointestinales
Pancreatitis autoinmune
La afectación pancreática constituye la manifestación más frecuente y mejor caracterizada de la IgG4-RD[4][5],[12],[13]. La pancreatitis autoinmune tipo 1 corresponde a la variante relacionada con IgG4, mientras que la tipo 2 no forma parte de este espectro[13],[14]. La forma tipo 1 se caracteriza por infiltrado linfoplasmocítico rico en células IgG4+, fibrosis periductal y esclerosis progresiva[13]-[15].
Clínicamente puede presentarse como ictericia obstructiva indolora, masa pancreática focal, agrandamiento difuso pancreático o, menos frecuentemente, episodios de pancreatitis aguda[13]-[15]. En fases avanzadas puede evolucionar hacia insuficiencia pancreática endocrina o exocrina[13].[14]. Desde el punto de vista radiológico, la forma difusa suele mostrar el clásico "páncreas en salchicha" (Figura 4), mientras que la forma focal puede simular un adenocarcinoma pancreático (Figura 5)[5],[14],[16]. El signo del halo capsular es otro hallazgo de imagen clásico y puede apoyar el diagnóstico (Figura 6), aunque no es patognomónico[5],[14],[16].
Figura 4
Corte coronal de TC de abdomen en el que se observa un aumento difuso de la glándula pancreática sin cambios inflamatorios peripancreáticos, compatible con “Páncreas en forma de salchicha”.
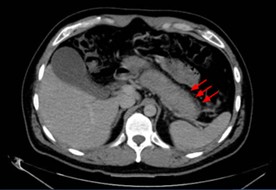
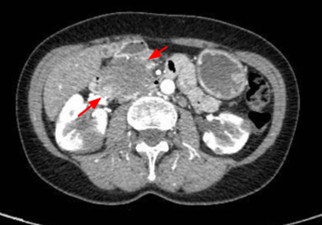
Figura 5
Corte transversal de TC de abdomen donde se objetiva una afectación focal en cabeza de páncreas (Flechas rojas) en contexto de PAI. Es necesario descartar malignidad.
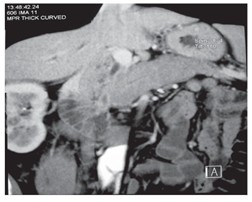
Figura 6
Corte transversal de TC de abdomen que muestra (Flechas rojas) un anillo periférico hipodenso que rodea a la glándula pancreática, conocido como el “signo del halo”.
El diagnóstico diferencial con el cáncer de páncreas es prioritario. La integración de datos clínicos, serológicos, radiológicos, histológicos y de afectación extrapancreática permite aumentar la precisión diagnóstica y evitar cirugías innecesarias[2],[13]-[16].
Afectación hepática y biliar
La manifestación biliar más relevante es la colangitis esclerosante asociada a IgG4, que aparece con frecuencia en pacientes con pancreatitis autoinmune tipo 1[4],[5],[17],[23]. Suele manifestarse como ictericia obstructiva o alteración analítica del perfil hepático[5],[17]. Las pruebas de imagen muestran estenosis largas y de contornos suaves, a diferencia del patrón típico de la colangitis esclerosante primaria; la TC y la colangio-RM permiten demostrar dilatación biliar y segmentos estenóticos característicos (Figura 7)[5],[17],[23]. La respuesta a glucocorticoides constituye un dato clínico muy orientador[17],[23].
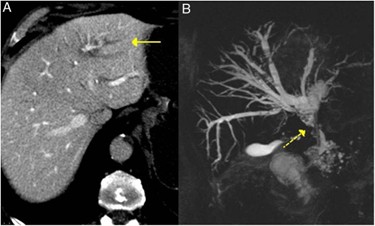
Figura 7
Imagen A muestra corte de TC con una dilatación de la vía biliar intrahepática izquierda (Flecha continua). Imagen B muestra corte de ColangioRM con dilatación de la vía biliar intrahepática izquierda y derecha y de la extrahepática, con estenosis en el tercio medio del colédoco (Flecha discontinua).
La hepatitis autoinmune asociada a IgG4 sigue siendo un área controvertida. Aunque se ha descrito un subgrupo de pacientes con hepatitis autoinmune clásica y abundante infiltrado portal de células plasmáticas IgG4+, todavía no está completamente aclarado si se trata de una verdadera manifestación hepática de la IgG4-RD o de un subtipo de hepatitis autoinmune[18]-[20].
Los pseudotumores hepáticos constituyen una forma menos frecuente de presentación. Histológicamente pueden mostrar un patrón fibroinflamatorio o linfoplasmocítico, y es esencial diferenciarlos de neoplasias primarias o metastásicas[4],[12].
Afectación del tracto gastrointestinal
La afectación directa del tubo digestivo es infrecuente y continúa siendo uno de los aspectos más controvertidos del espectro de la enfermedad[4],[6],[7],[12]. En la práctica clínica, la enfermedad digestiva relacionada con IgG4 se concentra fundamentalmente en el páncreas y la vía biliar, mientras que la afectación primaria del resto del tracto gastrointestinal parece mucho menos frecuente y está sustentada por una evidencia de menor solidez, basada en gran medida en casos aislados o series cortas. Esta limitación obliga a interpretar con cautela muchos de los hallazgos publicados y a evitar sobrediagnósticos en lesiones inflamatorias con infiltrado rico en células IgG4+ pero sin el contexto clínico, radiológico e histológico característico de la enfermedad[4],[6],[7],[12].
Se han descrito casos en esófago, estómago, intestino delgado, colon y papila mayor[4],[6],[21]. En muchos de ellos existe infiltración por células plasmáticas IgG4+ en mucosa o submucosa, aunque no siempre se identifican todos los criterios histológicos clásicos de la IgG4-RD, lo que ha alimentado el debate sobre si se trata de auténtica afectación gastrointestinal o de un fenómeno asociado[6],[7],[12].
En el esófago se han comunicado casos de disfagia, odinofagia, estenosis o masa pseudotumoral[4],[6]. En el estómago, la enfermedad puede manifestarse como ulceración, engrosamiento parietal difuso, pólipos o lesiones seudotumorales[6],[21]. En intestino delgado y colon, la evidencia disponible se basa principalmente en casos aislados o series cortas[4],[6],[12]. La afectación de la papila mayor, por su relativa frecuencia en pacientes con pancreatitis autoinmune, puede aportar información diagnóstica adicional[4],[12].
Mesenteritis esclerosante
La mesenteritis esclerosante asociada a IgG4 representa una manifestación poco frecuente pero clínicamente relevante[4],[22]. Puede presentarse con dolor abdominal, masa palpable, distensión, pérdida ponderal o incluso obstrucción intestinal[4],[22]. Las pruebas de imagen muestran masas mesentéricas, retracción fibrótica y adenopatías, y el diagnóstico diferencial incluye linfoma, carcinomatosis peritoneal, mesotelioma, sarcoidosis y enfermedad de Crohn[4],[22]. La demostración histológica de un infiltrado compatible y la respuesta al tratamiento ayudan a establecer el diagnóstico[4],[22].
Tratamiento
El tratamiento debe individualizarse según los órganos afectados, la gravedad clínica, el riesgo de daño irreversible y la probabilidad de recaída[2],[23],[27]. Los objetivos son inducir remisión, prevenir fibrosis progresiva y reducir la exposición acumulada a glucocorticoides [2],[23],[27].
Inducción de la remisión
Los glucocorticoides siguen siendo el tratamiento de primera línea en la mayoría de los pacientes[2],[13],[23]. Habitualmente se emplea prednisona a dosis de 0,5-1 mg/kg/día, mantenida durante 2-4 semanas y seguida de descenso gradual[2],[23]. La respuesta suele ser rápida tanto desde el punto de vista clínico como radiológico, especialmente en la pancreatitis autoinmune y en la afectación biliar[13],[17],[23].
Rituximab, anticuerpo monoclonal anti-CD20, ha demostrado elevada eficacia tanto en la inducción como en el mantenimiento de la remisión[2],[24],[25],[27]. Aunque inicialmente se reservó para casos refractarios o con recaídas frecuentes, su utilización como tratamiento de primera línea en pacientes seleccionados está ganando aceptación, especialmente en enfermedad multiorgánica grave, alto riesgo de recaída o contraindicación para glucocorticoides[2],[24],[27]. Los estudios disponibles han mostrado tasas altas de remisión sostenida y una reducción significativa de la exposición acumulada a corticoides[24],[25],[27]. No obstante, persisten incertidumbres relevantes sobre el mejor esquema de mantenimiento con rituximab, la duración óptima del tratamiento y la selección ideal de pacientes para retratamiento programado frente a retratamiento a demanda, por lo que su uso debe individualizarse según el perfil clínico y evolutivo de cada paciente[30].
En los últimos años, el desarrollo de terapias dirigidas frente a células B ha ampliado el horizonte terapéutico. En este contexto, inebilizumab, anticuerpo monoclonal anti-CD19, representa la novedad terapéutica más relevante. El ensayo fase 3 MITIGATE mostró que inebilizumab redujo el riesgo de brotes de la enfermedad y aumentó la probabilidad de remisión completa libre de brotes al año, confirmando el papel de la depleción de células B dirigidas frente a CD19 como una estrategia eficaz en la IgG4-RD[29]. Este hallazgo refuerza la idea de que el control de la enfermedad puede beneficiarse de abordajes dirigidos a linajes B más amplios que los alcanzados por el bloqueo anti-CD20 y apunta a un posible cambio de paradigma en pacientes con enfermedad recidivante, multiorgánica o dependiente de glucocorticoides[29].
Se han utilizado también fármacos ahorradores de glucocorticoides como azatioprina, micofenolato, metotrexato o leflunomida, aunque la evidencia que respalda su beneficio es menos robusta y los resultados son variables [2][23],[27]. Otras estrategias emergentes, como belimumab, obexelimab, rilzabrutinib o los inhibidores de la vía JAK-STAT, siguen siendo prometedoras, pero por ahora disponen de una base de evidencia claramente inferior a la acumulada para rituximab e inebilizumab[30].
Mantenimiento
La elevada tasa de recaídas justifica considerar tratamiento de mantenimiento en pacientes seleccionados, especialmente en aquellos con debut a edad temprana, afectación multiorgánica, elevación persistente de IgG4, enfermedad pancreatobiliar extensa o antecedentes de recaída[23]-[25]. Las estrategias más empleadas incluyen glucocorticoides a bajas dosis, rituximab periódico y, en determinados casos, inmunomoduladores como terapia ahorradora de glucocorticoides[23]-[25],[27]. Aun así, la estrategia óptima de mantenimiento continúa sin estar completamente definida y probablemente deba individualizarse según los órganos afectados, la gravedad inicial, la toxicidad acumulada, la respuesta previa al tratamiento y el riesgo estimado de recaída[24][25],[30].
Monitorización
La monitorización debe ser individualizada y combinar evaluación clínica, pruebas analíticas e imagen según la localización de la enfermedad[2],[23],[26][27]. La determinación seriada de IgG4 sérica puede ser útil en algunos pacientes, aunque su valor es limitado como marcador exclusivo de actividad[10],[26]. Los plasmablastos circulantes y otros biomarcadores emergentes podrían mejorar la monitorización en el futuro[10],[26],[27]. Dada la frecuencia de recaídas subclínicas, se recomienda reevaluación periódica incluso en ausencia de síntomas. Un esquema práctico de seguimiento clínico, analítico y radiológico puede verse resumido en la monitorización propuesta por los autores (Figura 8)[23],[25],[27].
Asimismo se propone un cuadro resumen donde se incluye el manejo diagnóstico y terapéutico de la enfermedad gastrointestinal asociada a la IgG4 (Figura 9).
Tabla 1
Cuadro resumen sobre el manejo diagnóstico y terapéutico de la Enfermedad Gastrointestinal asociada a IgG4. Elaborado por el autor.
Pronóstico y perspectivas futuras
El pronóstico suele ser favorable cuando el diagnóstico se establece de forma precoz y se instaura tratamiento adecuado[2],[27]. No obstante, la fibrosis establecida puede dejar secuelas permanentes y condicionar disfunción orgánica persistente. La recaída constituye uno de los principales problemas a largo plazo, especialmente en la afectación pancreatobiliar[23]-[25],[27].
La asociación entre IgG4-RD y malignidad continúa siendo motivo de debate. Aunque la causalidad no está demostrada, trabajos recientes siguen sugiriendo un aumento del riesgo global de cáncer y de determinados tumores concretos en pacientes con IgG4-RD, con especial atención a neoplasias pancreatobiliares y algunas malignidades hematológicas, así como un posible pico de riesgo en los primeros meses o durante el primer año tras el diagnóstico de la enfermedad[28],[31],[32]. Este patrón obliga a una vigilancia clínica cuidadosa y a un despistaje razonable de procesos neoplásicos cuando la presentación clínica lo sugiera, aunque la evidencia actual no justifica, por sí sola, estrategias de cribado oncológico distintas de las individualizadas según la edad, los factores de riesgo y el contexto clínico de cada paciente[31],[32].
La mejor comprensión de la inmunopatogenia está impulsando el desarrollo de terapias más específicas y estrategias de medicina personalizada. En este sentido, la identificación de biomarcadores dinámicos de actividad y el avance de tratamientos dirigidos contra células B, plasmablastos y vías de señalización inmunitaria representan líneas de investigación especialmente prometedoras [2][10],[26],[27],[29],[30]. La incorporación de inebilizumab al escenario terapéutico y el desarrollo de otros agentes dirigidos refuerzan la idea de que el abordaje de la IgG4-RD podría evolucionar en los próximos años hacia modelos más estratificados según riesgo de recaída, patrón de afectación orgánica y perfil inmunobiológico del paciente[29],[30].
Conclusiones
La enfermedad relacionada con IgG4 es una patología fibroinflamatoria sistémica de gran relevancia para el especialista en Aparato Digestivo por su frecuente afectación pancreatobiliar y por su capacidad para simular procesos neoplásicos[2],[4],[5],[23]. Su diagnóstico exige una integración rigurosa de hallazgos clínicos, radiológicos, serológicos e histológicos, evitando interpretaciones aisladas de la IgG4 sérica o de la respuesta terapéutica[2][3],[8],[26].
Los glucocorticoides continúan siendo la base del tratamiento inicial, mientras que rituximab se ha consolidado como una opción eficaz en enfermedad refractaria, recidivante o de alto riesgo, aunque persisten dudas sobre la estrategia óptima de mantenimiento[24],[25],[30]. La irrupción de inebilizumab, respaldada por evidencia positiva de fase 3, refuerza el papel de las terapias dirigidas contra células B y apunta a una nueva etapa en el manejo de la enfermedad[29]. La expansión del conocimiento sobre mecanismos inmunológicos y biomarcadores, junto con el desarrollo de nuevas terapias dirigidas, probablemente transformará el abordaje de esta entidad en los próximos años[2],[10],[26],[27],[29],[30].