
 Descargar número completo
Descargar número completo Download full issue
Download full issueCITE THIS WORK
Fernandez Carrasco M, Plaza Fernández A, Rodríguez Mateu A. Richter's syndrome as a manifestation of intestinal lymphoma: a rare form of aggressive transformation of chronic lymphocytic leukemia. RAPD 2026;49(2):59-61. DOI: 10.37352/2026492.3
Introduction
RS represents one of the most serious complications of CLL and is characterized by transformation to an aggressive lymphoma, predominantly DLBCL. Its annual incidence is around 0.5–1%, and the cumulative risk throughout the course of the disease has been estimated at between 2–10%. The usual presentation includes accelerated adenopathic growth, B symptoms, and marked elevation of LDH, with median survival generally less than one year. Gastrointestinal tract (GIT) involvement is rare and can make diagnosis difficult by mimicking primary digestive neoplasms, inflammatory bowel disease, or infectious processes.[1]-[3].
Clinical case
A 54-year-old man with a history of Hodgkin's disease treated with chemotherapy in 2003. In 2021, he was diagnosed with B-CLL, initially stage 0. In 2022, in the presence of lymphocyte duplication, 13q and 17p deletions were detected, and treatment with ibrutinib was initiated.
In January 2025, he was admitted for abdominal pain, asthenia, and weight loss, with severe anemia, lymphocytosis, and splenomegaly. Laboratory tests showed hemoglobin of 7.4 g/dL, LDH of 1,120 U/L, beta-2 microglobulin of 6.8 mg/L, and corrected serum calcium of 11.3 mg/dL.
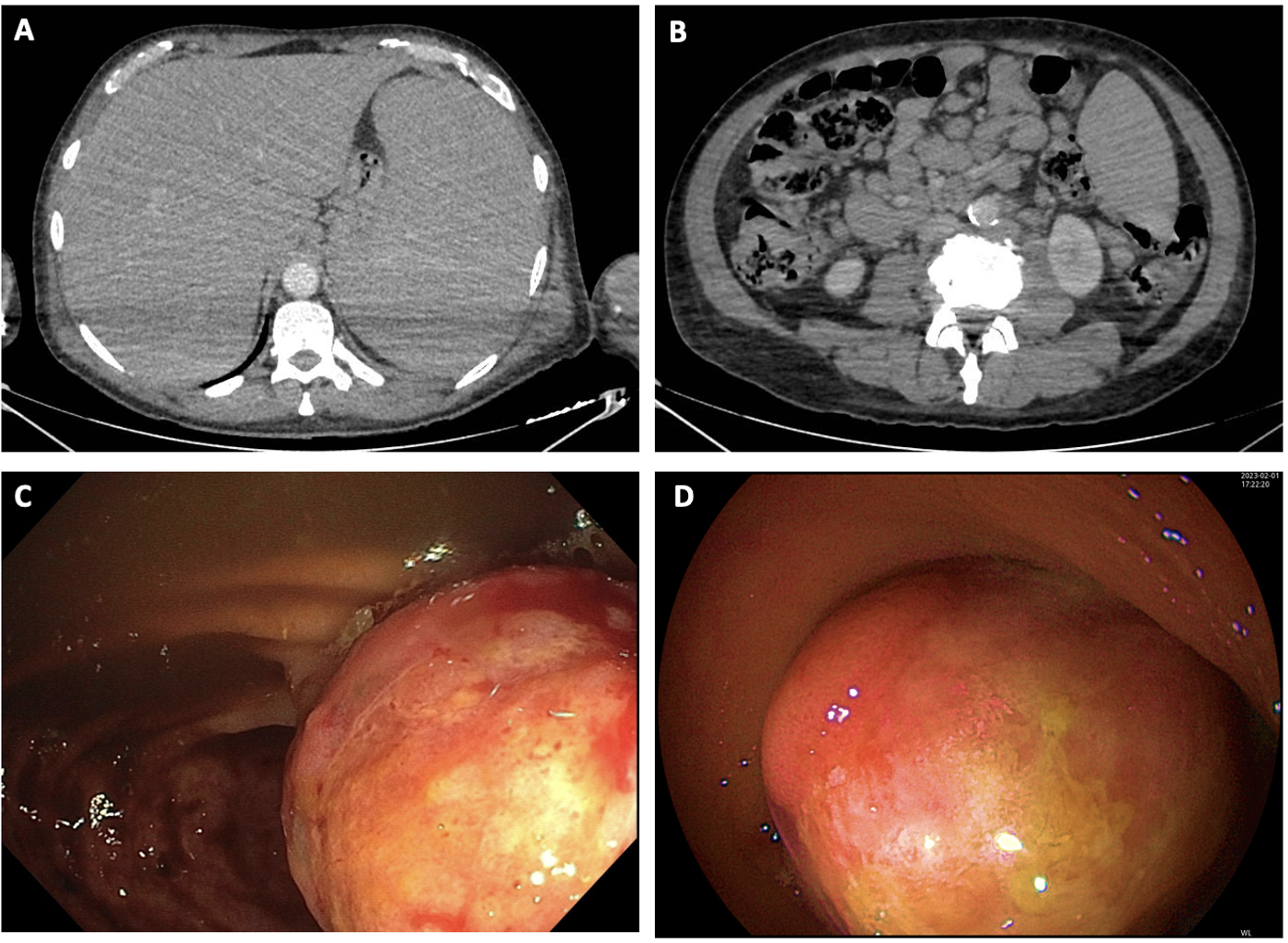
PET-CT revealed hypermetabolic lymphadenopathy, splenomegaly, and nonspecific thickening in the right colon and cecum. Colonoscopy was performed, revealing a thickened and ulcerated ileocecal valve with an infiltrative appearance. Biopsy confirmed infiltration by lymphoid neoplasia.
The histological study was consistent with RS due to transformation from CLL to DLBCL, non-germinal center variant. Immunohistochemistry showed CD20+, CD5+, CD23+, BCL2+, MUM-1+ B cells, with a Ki-67 proliferative index of 35–40%, with no evidence of Epstein–Barr virus infection. The conclusion was aggressive transformation with lymphatic and splenic dissemination, with fulminant progression and fatal outcome.
Discussion
RS is a clinically heterogeneous entity with a poor prognosis. Around 90% of transformations correspond to DLBCL, while the Hodgkin lymphoma variant accounts for approximately 5–10%. Biological aggressiveness is often associated with high-risk genetic alterations, including TP53, NOTCH1, and MYC, which are implicated in therapeutic resistance and lower survival. The presence of 17p deletion in our patient, widely linked to TP53 dysfunction, is an unfavorable prognostic marker and may contribute to rapid clinical progression[1],[2].
Gastrointestinal involvement by RS is rare and is usually described as infiltrative or ulcerative lesions in the terminal ileum, ileocecal valve, or colon, with symptoms such as abdominal pain, anemia, weight loss, or even gastrointestinal bleeding. Its clinical significance lies in the fact that it can be confused with primary digestive pathology, delaying diagnosis. In these cases, the combination of systemic symptoms (B symptoms), suggestive laboratory data (elevated LDH, anemia, occasional hypercalcemia), and imaging findings can guide suspicion.
PET-CT is particularly useful for locating areas of high metabolic activity and guiding the collection of biopsies from the most representative tissue. In our case, the correlation between hypermetabolic lymphadenopathy and colonic thickening guided the endoscopic study and confirmed the diagnosis[3].
The treatment of RS is based on R-CHOP or R-EPOCH chemoimmunotherapy regimens, with generally limited responses. In selected patients, combinations with venetoclax, anti-PD1 blockade immunotherapy, and cellular strategies such as CAR-T have been evaluated, especially in refractory or high-risk disease. Even so, overall survival remains poor, reinforcing the importance of early clinical suspicion[4].
In the present case, the fast progression, clinical deterioration, and aggressiveness of the disease resulted in a very limited therapeutic range, making it difficult to initiate intensive treatment. This highlights that gastrointestinal involvement can act as a marker of aggressiveness and dissemination, and underscores the need for rapid diagnosis in the event of atypical digestive manifestations in patients with CLL.
Conclusion
RS is a rare but highly aggressive complication of CLL. Although gastrointestinal involvement is rare, it should be considered in the presence of abdominal symptoms associated with systemic clinical manifestations or suggestive findings on PET-CT. Endoscopy with targeted biopsy is essential to confirm the diagnosis. Given the poor prognosis, especially in the presence of high-risk alterations such as 17p/TP53 deletion, early recognition may be decisive in planning therapeutic strategies before irreversible clinical deterioration occurs.