
 Descargar número completo
Descargar número completo Download full issue
Download full issueCITE THIS WORK
López Ortega S, Parra López B, Alonso Belmonte C, González Grande R, Jiménez Pérez M. IgG4-related gastrointestinal disease. RAPD 2026;49(2):50-58. DOI: 10.37352/2026492.2
Introduction
IgG4-related disease (IgG4-RD) was recognized as a distinct entity at the beginning of this century following the description of elevated serum IgG4 levels in patients with sclerosing pancreatitis[1]. It is currently considered a systemic fibroinflammatory disease capable of affecting numerous organs, including the pancreas, biliary tract, liver, salivary glands, kidneys, retroperitoneum, lungs, and gastrointestinal tract[2],[3],[8]. Its clinical significance lies in its ability to mimic neoplasms or other inflammatory diseases and in the risk of irreversible fibrosis when diagnosis and treatment are delayed[2],[8],[27].
From a gastrointestinal perspective, type 1 autoimmune pancreatitis is the most common manifestation, but not the only one. IgG4-associated sclerosing cholangitis, liver involvement, gastrointestinal tract infiltration, and sclerosing mesenteritis broaden the clinical spectrum and present significant diagnostic challenges for the gastroenterologist[4]-[7],[12].
The aim of this review is to provide an update on the epidemiological, pathogenic, diagnostic, and therapeutic aspects of IgG4-RD, with an emphasis on gastrointestinal manifestations and the practical elements of greatest clinical utility.
IgG4-related disease
IgG4-RD is a systemic immune-mediated disease characterized by IgG4-positive plasma cell-rich lymphoplasmacytic infiltration, fibrosis, and, depending on the organ, obliterative phlebitis[2],[8],[11]. Its presentation is heterogeneous and depends on the anatomical location and the number of organs involved. More than half of patients have multiorgan involvement, and prolonged disease progression can lead to irreversible structural damage[8],[12],[27].
Epidemiology
The disease predominantly affects adults between the ages of 50 and 70, although pediatric and elderly cases have been reported[5],[7]. There is a male predominance in the pancreatobiliary and retroperitoneal forms, while head and neck involvement may be relatively more common in women[5],[8]. The incidence and prevalence are likely underdiagnosed. Recent population-based studies in the United States estimate an approximate incidence of 0.78–1.39 cases per 100,000 person-years and a rising prevalence, in parallel with increased recognition of the disease[9].
Pathogenesis
The pathogenesis of IgG4-RD is complex and involves a combination of genetic susceptibility, repeated antigenic exposure, and a dysregulated immune response[2],[8],[10]. Key mechanisms include the activation of follicular T helper cells, which promote isotype switching in B cells toward IgG4 and IgE, and the interaction between B cells, plasmablasts, and effector T cell subpopulations in target tissues[8],[10].
Recent studies have established the central role of clonal cytotoxic CD4+ T cells, particularly those with the SLAMF7+ phenotype, as the primary effectors of tissue damage[10],[27]. These cells produce profibrotic cytokines such as TGF-β and IFN-γ, exhibit clonal expansion in peripheral blood and affected tissues, and correlate with disease activity[10],[27]. It has also been suggested that part of the therapeutic effect of rituximab may depend on the disruption of the antigenic support that B cells provide to these effector T cell populations[2],[10],[27].
Likewise, circulating plasmablasts have emerged as a dynamic biomarker of inflammatory activity. Their quantification appears to offer greater sensitivity than serum IgG4 in some contexts, including in patients with normal levels of this immunoglobulin, which reinforces their potential utility for monitoring[10],[26],[27].
Despite the association between the disease and elevated serum and tissue IgG4 levels, the exact role of IgG4 in the pathogenesis remains unclear. Rather than necessarily acting as a direct mediator of tissue damage, it may serve as a marker of a more complex abnormal immune response[8],[10].
Histopathology
Histopathology is a cornerstone of diagnosis. The three classic features are: dense lymphoplasmacytic infiltrate, storiform fibrosis, and obliterative phlebitis[8],[11]. The expression of each of these features varies depending on the affected organ, and they are not always present simultaneously. The diffuse lymphoplasmacytic infiltrate represents one of the most characteristic findings on histological examination (Figure 1), while storiform fibrosis constitutes another of the most representative morphological features of the condition (Figure 2)[8],[11]. Obliterative phlebitis, when present, also carries significant diagnostic weight (Figure 3)[8],[11]. Immunohistochemistry demonstrates an increase in IgG4+ plasma cells and an elevated IgG4+/IgG+ ratio, typically exceeding 40%, although the cutoff value should be interpreted according to the tissue analyzed[8],[11].
Figura 1
Histological image showing swirling fibrotic tissue surrounding a cellular infiltrate within collagen fibrils (A). Image B shows stellate fibrosis surrounded by a lymphoplasmacytic infiltrate.
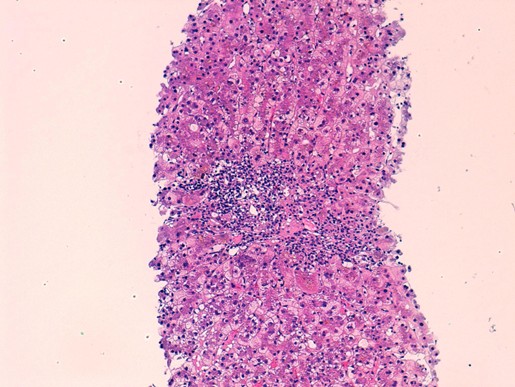
Figura 2
Histological image showing a dense, diffuse lymphoplasmacytic infiltrate. Courtesy of Dr. Teresa Pereda, Costa del Sol Hospital (Marbella).
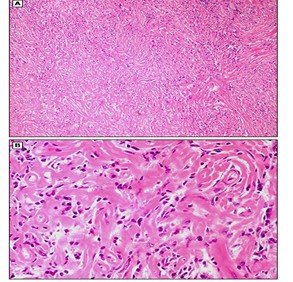
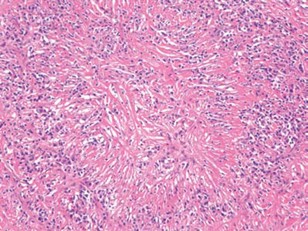
Figura 3
Histological image showing partial or complete obliteration of the medium-sized veins, secondary to lymphoplasmacytic infiltration, consistent with obliterative phlebitis.
Storiform fibrosis represents a whorled arrangement of collagen bundles, particularly useful for diagnosis in deep organs[11]. It should be noted that the patchy distribution of lesions may limit the yield of small biopsies, especially in needle-core samples[11],[15].
Serology and biomarkers
Most patients have elevated serum IgG4 levels, although these may be normal in a significant percentage of cases[5],[8],[26]. Furthermore, elevated serum IgG4 is nonspecific and can be observed in other inflammatory, infectious, or neoplastic diseases[5],[8],[26]. Therefore, its utility is complementary and never sufficient to confirm the diagnosis on its own.
In addition to serum IgG4, other immunological abnormalities of interest have been described. Peripheral eosinophilia and elevated serum IgE are relatively common findings and reflect a predominantly Th2 immune profile[10],[26]. In patients with renal involvement, hypocomplementemia may be observed, with decreased C3 and C4 levels, associated with increased inflammatory activity[26]. However, there is currently no single biomarker with sufficient sensitivity and specificity to establish a diagnosis of IgG4-RD on its own[2],[10],[26].
Clinical presentation
The clinical presentation of the disease is highly variable and often insidious[2],[5],[8]. General symptoms include asthenia, weight loss, low-grade fever, pain or enlargement of the affected organ, and, in some cases, systemic symptoms[5],[8],[12]. This nonspecific profile necessitates a broad differential diagnosis, which includes neoplasms, lymphomas, infections, and other autoimmune or fibroinflammatory diseases[2],[8][27].
Imaging tests and classification criteria
Imaging techniques play a fundamental role in both diagnosis and in assessing extent and monitoring[4][5],[13],[16]. Contrast-enhanced computed tomography (CT) is useful in the initial evaluation, especially of the pancreas, biliary tract, retroperitoneum, and large vessels[4],[5],[16]. Magnetic resonance imaging (MRI) and magnetic resonance cholangiography (MRCP) provide better tissue and ductal characterization[4],[5],[16]. Positron emission tomography (PET-CT) can identify subclinical multiorgan involvement and is particularly valuable in systemic disease[2],[4]. Endoscopic ultrasound allows for targeted tissue sampling in the pancreas and biliary tract[4],[13].
In 2019, the American College of Rheumatology and the European League Against Rheumatism developed classification criteria with the aim of standardizing patient inclusion in clinical studies[3]. These criteria were designed for classification purposes rather than strictly for diagnosis; therefore, their application in clinical practice must be contextualized within a comprehensive evaluation[2],[3]. The diagnosis of IgG4-RD continues to be based on a combination of clinical, serological, radiological, and histopathological findings, with the exclusion of mimicking conditions[2],[3],[8]).
General diagnosis
In the context of pancreatic involvement, the HISORt criteria remain highly clinically useful, especially when histological confirmation is not feasible[13],[15]. This approach integrates histology, imaging, serology, involvement of other organs, and response to treatment[13],[15]. Response to glucocorticoids, in the absence of data suggestive of malignancy, remains a relevant guiding factor, although it should never be used in isolation if suspicion of cancer persists[2],[13],[15].
Gastrointestinal manifestations
Autoimmune Pancreatitis
Pancreatic involvement is the most common and best-characterized manifestation of IgG4-related disease[4][5],[12],[13]. Type 1 autoimmune pancreatitis corresponds to the IgG4-related variant, whereas type 2 does not fall within this spectrum[13],[14]. The type 1 form is characterized by an IgG4+ cell-rich lymphoplasmacytic infiltrate, periductal fibrosis, and progressive sclerosis[13]-[15].
Clinically, it may present as painless obstructive jaundice, a focal pancreatic mass, diffuse pancreatic enlargement, or, less frequently, episodes of acute pancreatitis[13]-[15]. In advanced stages, it may progress to endocrine or exocrine pancreatic insufficiency[13].[14]. From a radiological perspective, the diffuse form typically shows the classic "sausage-shaped pancreas"(Figure 4), while the focal form may mimic pancreatic adenocarcinoma (Figure 5)[5],[14],[16]. The capsular halo sign is another classic imaging finding and may support the diagnosis(Figure 6), although it is not pathognomonic[5],[14],[16].
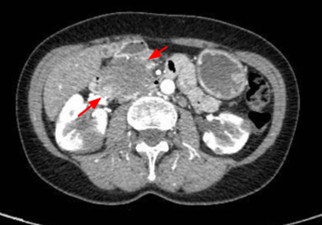
Figura 4
A cross-sectional CT scan of the abdomen showing (red arrows) a hypodense peripheral ring surrounding the pancreas, known as the “halo sign.”
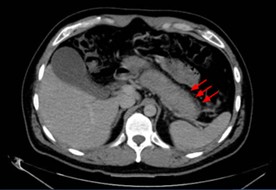
Figura 5
Coronal CT scan of the abdomen showing diffuse enlargement of the pancreas without peripancreatic inflammatory changes, consistent with “sausage-shaped pancreas.”
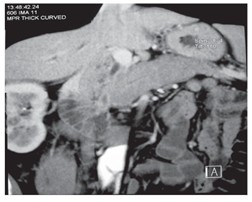
Figura 6
Abdominal CT cross-section showing focal involvement of the pancreatic head (red arrows) in the context of PAI. Malignancy must be ruled out.
Differential diagnosis with pancreatic cancer is a priority. Integrating clinical, serological, radiological, histological, and extrapancreatic data helps improve diagnostic accuracy and avoid unnecessary surgeries[2],[13]-[16].
Hepatic and biliary involvement
The most significant biliary manifestation is IgG4-associated sclerosing cholangitis, which frequently occurs in patients with type 1 autoimmune pancreatitis[4],[5],[17],[23]. It typically presents as obstructive jaundice or abnormal liver function tests[5],[17]. Imaging studies show long, smoothly contoured strictures, unlike the typical pattern of primary sclerosing cholangitis; CT and MR cholangiography demonstrate biliary dilation and characteristic stenotic segments (Figure 7)[5],[17],[23]. Response to glucocorticoids is a highly informative clinical finding[17],[23].
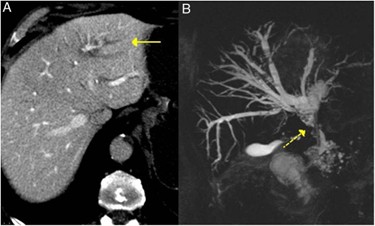
Figura 7
Image A shows a CT slice with dilation of the left intrahepatic bile duct (solid arrow). Image B shows an MR cholangiography slice with dilation of the left and right intrahepatic bile ducts and the extrahepatic bile duct, with stenosis in the middle third of the common bile duct (dashed arrow).
IgG4-associated autoimmune hepatitis remains a controversial area. Although a subgroup of patients with classic autoimmune hepatitis and abundant portal infiltration of IgG4+ plasma cells has been described, it is not yet fully clarified whether this represents a true hepatic manifestation of IgG4-RD or a subtype of autoimmune hepatitis[18]-[20].
Hepatic pseudotumors represent a less common form of presentation. Histologically, they may show a fibroinflammatory or lymphoplasmacytic pattern, and it is essential to differentiate them from primary or metastatic neoplasms[4],[12].
Involvement of the gastrointestinal tract
Direct involvement of the gastrointestinal tract is rare and remains one of the most controversial aspects of the disease spectrum[4],[6],[7],[12]. In clinical practice, IgG4-related gastrointestinal disease is primarily confined to the pancreas and biliary tract, whereas primary involvement of the rest of the gastrointestinal tract appears much less common and is supported by less robust evidence, based largely on isolated cases or small series. This limitation necessitates cautious interpretation of many published findings and the avoidance of overdiagnosis in inflammatory lesions with an IgG4+ cell-rich infiltrate but without the clinical, radiological, and histological features characteristic of the disease[4],[6],[7],[12].
Cases have been described in the esophagus, stomach, small intestine, colon, and major papilla[4],[6],[21]. In many of these cases, there is infiltration by IgG4+ plasma cells in the mucosa or submucosa, although not all classic histological criteria for IgG4-RD are always identified, which has fueled the debate over whether this represents true gastrointestinal involvement or an associated phenomenon[6],[7],[12].
Cases of dysphagia, odynophagia, stenosis, or pseudotumoral masses have been reported in the esophagus[4],[6]. In the stomach, the disease may present as ulceration, diffuse parietal thickening, polyps, or pseudotumoral lesions[6],[21]. In the small intestine and colon, the available evidence is based primarily on isolated cases or short series[4],[6],[12]. Involvement of the major papilla, due to its relative frequency in patients with autoimmune pancreatitis, may provide additional diagnostic information[4],[12].
Sclerosing mesenteritis
IgG4-associated sclerosing mesenteritis represents a rare but clinically relevant manifestation[4],[22]. It may present with abdominal pain, a palpable mass, distension, weight loss, or even intestinal obstruction[4],[22]. Imaging studies show mesenteric masses, fibrotic retraction, and lymphadenopathy, and the differential diagnosis includes lymphoma, peritoneal carcinomatosis, mesothelioma, sarcoidosis, and Crohn’s disease[4],[22]. Histological demonstration of a compatible infiltrate and response to treatment help establish the diagnosis[4],[22].
Treatment
Treatment should be personalised according to the affected organs, clinical severity, risk of irreversible damage, and likelihood of relapse[2],[23],[27]. The goals are to induce remission, prevent progressive fibrosis, and reduce cumulative exposure to glucocorticoids[2],[23],[27].
Induction of remission
Glucocorticoids remain the first-line treatment in most patients[2],[13],[23]. Prednisone is typically used at a dose of 0.5–1 mg/kg/day, maintained for 2–4 weeks and followed by gradual tapering[2],[23]. The response is usually rapid from both a clinical and radiological perspective, especially in autoimmune pancreatitis and biliary involvement[13],[17],[23].
Rituximab, an anti-CD20 monoclonal antibody, has demonstrated high efficacy in both inducing and maintaining remission[2],[24],[25],[27]. Although initially reserved for refractory cases or those with frequent relapses, its use as first-line therapy in selected patients is gaining acceptance, especially in severe multiorgan disease, high risk of relapse, or contraindications to glucocorticoids[2],[24],[27]. Available studies have shown high rates of sustained remission and a significant reduction in cumulative corticosteroid exposure[24],[25],[27]. However, significant uncertainties remain regarding the best rituximab maintenance regimen, the optimal duration of treatment, and the ideal selection of patients for scheduled versus on-demand retreatment; therefore, its use should be individualised according to each patient’s clinical profile and disease progression[30].
In recent years, the development of B-cell-targeted therapies has broadened the therapeutic landscape. In this context, inebilizumab, an anti-CD19 monoclonal antibody, represents the most significant therapeutic advancement. The phase 3 MITIGATE trial showed that inebilizumab reduced the risk of disease flare-ups and increased the probability of complete remission free of flare-ups at one year, confirming the role of CD19-targeted B-cell depletion as an effective strategy in IgG4-RD[29]. This finding reinforces the idea that disease control may benefit from approaches targeting broader B-cell lineages than those reached by anti-CD20 blockade and points to a possible paradigm shift in patients with relapsing, multiorgan, or glucocorticoid-dependent disease[29].
Glucocorticoid-sparing drugs such as azathioprine, mycophenolate, methotrexate, or leflunomide have also been used, although the evidence supporting their benefit is less robust and results are variable[2][23],[27]. Other emerging strategies, such as belimumab, obexelimab, rilzabrutinib, or JAK-STAT pathway inhibitors, remain promising, but for now they have a clearly weaker evidence base than that accumulated for rituximab and inebilizumab[30].
Maintenance
The high relapse rate justifies considering maintenance therapy in selected patients, especially those with early-onset disease, multiorgan involvement, persistent elevated IgG4 levels, extensive pancreatobiliary disease, or a history of relapse[23]-[25]. The most commonly used strategies include low-dose glucocorticoids, periodic rituximab, and, in certain cases, immunomodulators as glucocorticoid-sparing therapy[23]-[25],[27]. Nevertheless, the optimal maintenance strategy remains undefined and should likely be tailored to the affected organs, initial severity, cumulative toxicity, prior treatment response, and estimated risk of relapse[24][25],[30].
Monitoring
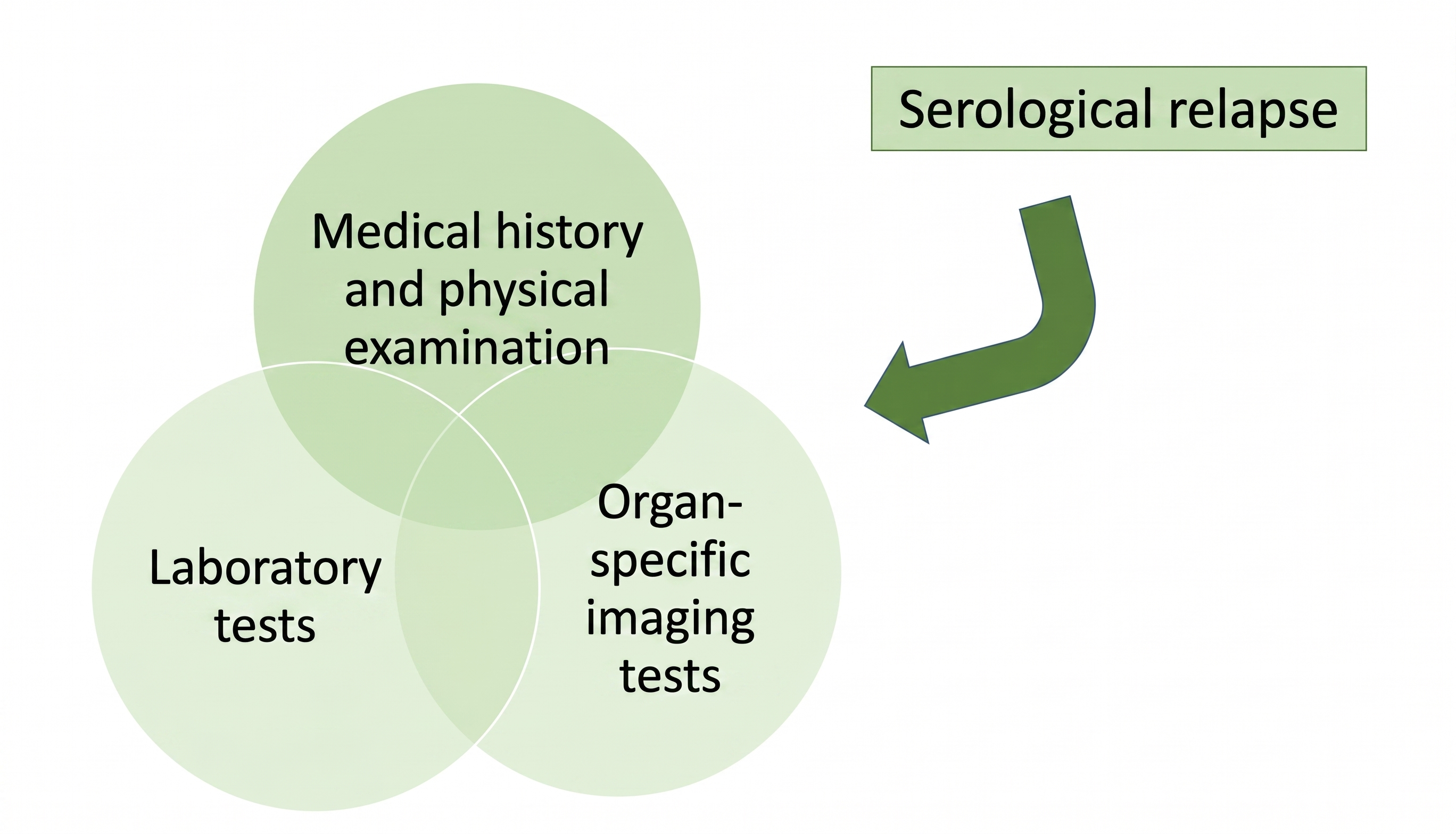
Monitoring should be personalised and combine clinical evaluation, laboratory tests, and imaging based on the location of the disease[2],[23],[26][27]. Serial measurement of serum IgG4 may be useful in some patients, although its value is limited as a sole marker of disease activity[10],[26]. Circulating plasmablasts and other emerging biomarkers could improve monitoring in the future[10],[26],[27]. Given the frequency of subclinical relapses, periodic reevaluation is recommended even in the absence of symptoms. A practical outline of clinical, laboratory, and radiological follow-up is summarized in the monitoring protocol proposed by the authors (Figure 8)[23],[25],[27].
A summary table is also provided, which includes the diagnostic and therapeutic management of IgG4-associated gastrointestinal disease (Figure 9).
Figura 9
Summary table on the diagnostic and therapeutic management of IgG4-associated gastrointestinal disease. Developed by the author.
Prognosis and future outlook
The prognosis is generally favorable when the diagnosis is made early and appropriate treatment is initiated[2],[27]. However, established fibrosis can lead to permanent sequelae and cause persistent organ dysfunction. Relapse is one of the main long-term problems, especially in pancreatobiliary involvement[23]-[25],[27].
The association between IgG4-RD and malignancy remains a subject of debate. Although causality has not been proven, recent studies continue to suggest an increased overall risk of cancer and of specific tumors in patients with IgG4-RD, with particular attention to pancreatobiliary neoplasms and certain hematologic malignancies, as well as a possible peak in risk during the first few months or the first year following diagnosis of the disease[28],[31],[32]. This pattern necessitates careful clinical monitoring and reasonable screening for neoplastic processes when the clinical presentation suggests it, although current evidence does not, on its own, justify cancer screening strategies other than those tailored to each patient’s age, risk factors, and clinical context[31],[32].
A better understanding of the immunopathogenesis is driving the development of more targeted therapies and personalized medicine strategies. In this regard, the identification of dynamic biomarkers of disease activity and advances in treatments targeting B cells, plasmablasts, and immune signaling pathways represent particularly promising lines of research[2][10],[26],[27],[29],[30]. The incorporation of inebilizumab into the therapeutic landscape and the development of other targeted agents reinforce the idea that the management of IgG4-RD could evolve in the coming years toward more stratified models based on relapse risk, pattern of organ involvement, and the patient’s immunobiological profile[29],[30].
Conclusions
IgG4-related disease is a systemic fibroinflammatory condition of great relevance to gastroenterologists due to its frequent involvement of the pancreas and biliary tract and its ability to mimic neoplastic processes[2],[4],[5],[23]. Its diagnosis requires a rigorous integration of clinical, radiological, serological, and histological findings, avoiding isolated interpretations of serum IgG4 or the therapeutic response[2][3],[8],[26].
Glucocorticoids remain the cornerstone of initial treatment, while rituximab has established itself as an effective option for refractory, relapsed, or high-risk disease, although questions remain regarding the optimal maintenance strategy[24],[25],[30]. The emergence of inebilizumab, supported by positive phase 3 evidence, reinforces the role of B-cell-targeted therapies and points to a new era in the management of the disease[29]. Expanding knowledge of immunological mechanisms and biomarkers, along with the development of new targeted therapies, will likely transform the approach to this condition in the coming years[2],[10],[26],[27],[29],[30].