
 Descargar número completo
Descargar número completo Download full issue
Download full issueCITE THIS WORK
de Vicente-Ortega A, Bracho-González M, Fernández-del Corral MR. Gastrointestinal amyloidosis as the initial manifestation of systemic amyloidosis. RAPD 2026;49(3):100-102. DOI: 10.37352/2026493.4
Introducción
Amyloidosis is a disease characterized by the extracellular deposition of fibrillar protein material, which leads to changes in the morphology and function of the tissue where it is deposited. Primary amyloidosis is a rare disease that affects multiple organs and has a poor prognosis[5].
Gastrointestinal (GI) involvement is rare and usually paucisymptomatic; it is rarely the initial presentation of the disease and is most frequently observed in the context of primary systemic amyloidosis[1],[4]. In the GI presentation, endoscopic findings are nonspecific[4]. The diagnosis is based on the demonstration of amyloid deposits in the tissues, and treatment must be individualized based on age, the degree, and type of organ involvement[5].
Clinical case
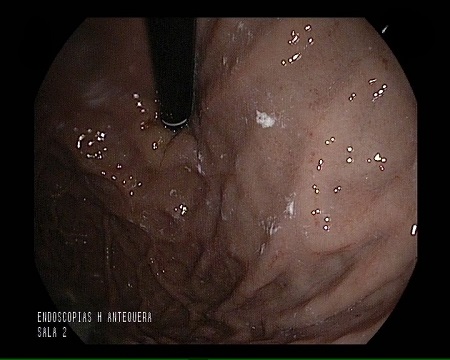
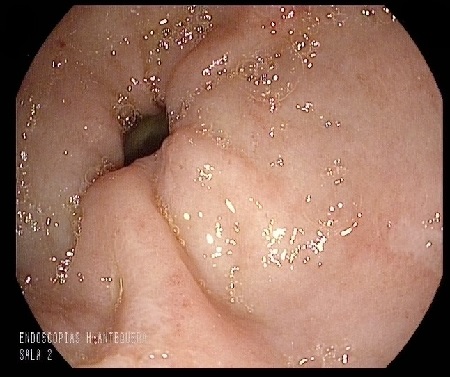
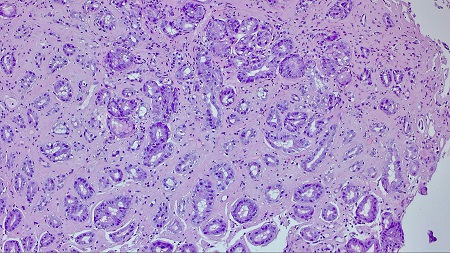
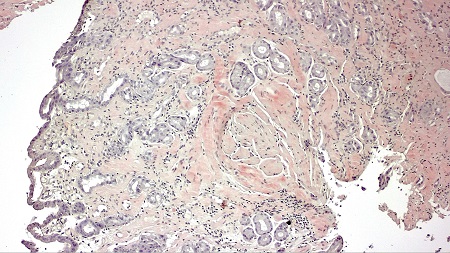
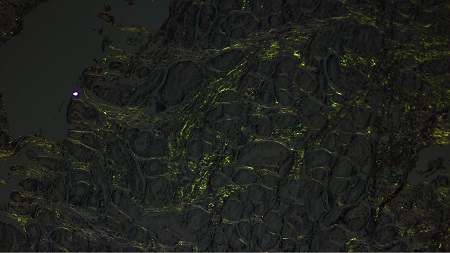
A 74-year-old man was admitted for abdominal pain, vomiting, constipation, and weight loss. During the workup, a gastroscopy was performed, revealing a gastric mucosa with an edematous, nodular appearance and several nonspecific erosions (Figures 1 and 2). With gastritis suspected, biopsies were taken; given the evidence of hyalinization in the lamina propria, the pathology department expanded the investigation. The Congo red histochemical technique was performed, revealing the presence of amorphous and hyaline material exhibiting birefringence, consistent with amyloid material (Figures 3-5). Based on these findings, a diagnosis of gastrointestinal amyloidosis was made.
During follow-up after discharge, the patient reported new symptoms including dyspnea related to decompensated heart failure, neurological problems, and the appearance of a skin lesion that was biopsied and diagnosed as primary systemic kappa-amyloidosis (AL), with follow-up and treatment provided by the Hematology Department.
Discussion
GI amyloidosis, and specifically gastric involvement, is a rare manifestation of primary systemic amyloidosis, occurring less frequently than renal and cardiac involvement[1],[3].
Gastrointestinal symptoms as the initial presentation are highly uncommon; however, in our case, the onset consisted of gastrointestinal symptoms. It is usually asymptomatic or nonspecific, but may present with symptoms such as: gastrointestinal bleeding, protein-losing gastroenteropathy, malabsorption, and motility disorders, the latter including: nausea, vomiting, gastroesophageal reflux, anorexia, constipation, chronic intestinal pseudo-obstruction, or gastroparesis[1],[2]. In our case, symptoms of motility disorders were present.
As was the case in our patient, endoscopic findings are nonspecific, with the main endoscopic findings being erosions, ulcerations, and nodular-appearing mucosa, as well as pseudopolypoid protrusions[4] ; diagnosis requires a biopsy to confirm the deposit using Congo red staining[5].