
 Descargar número completo
Descargar número completo Download full issue
Download full issueCP-212. CAUSA INUSUAL DE TRASPLANTE HEPÁTICO: POLINEUROPATÍA AMILOIDÓTICA FAMILIAR
Hallouch Toutouh, S. UGC de Aparato Digestivo. Complejo Hospitalario de Especialidades Torrecárdenas. Almería
Barrientos Delgado, A. UGC de Aparato Digestivo. Complejo Hospitalario de Especialidades Torrecárdenas. Almería
Jordán Madrid, T. UGC de Aparato Digestivo. Complejo Hospitalario de Especialidades Torrecárdenas. Almería
Delgado Maroto, A. UGC de Aparato Digestivo. Complejo Hospitalario de Especialidades Torrecárdenas. Almería
Práxedes González, E. UGC de Aparato Digestivo. Complejo Hospitalario de Especialidades Torrecárdenas. Almería
Casado Martin, M. UGC de Aparato Digestivo. Complejo Hospitalario de Especialidades Torrecárdenas. Almería
Vega Sáenz, JL. UGC de Aparato Digestivo. Complejo Hospitalario de Especialidades Torrecárdenas. Almería
Introducción
Bajo el título de las amiloidosis se engloba, un grupo heterogéneo de entidades cuyo nexo común es, el depósito patológico en distintos órganos y sistemas, de una proteína, alterando de esta manera, su correcto funcionamiento. A pesar de no ser la forma más frecuente de amiloidosis, la polineuropatía amiloidótica familiar (PAF), es una patología a ser considerada por el especialista en gastroenterología, al ser el trasplante hepático, su único tratamiento etiológico y tiene un curso inexorablemente progresivo causando la muerte 7-15 años después del inicio de las manifestaciones clínicas.
Material y métodos
Presentamos el caso clínico de un varón de 65 con antecedentes de hipertensión, dislipemia y bloqueo de rama derecha. Es derivado a consulta de digestivo, tras ser estudiado por el servicio de cardiología, al fallecer su hermano por amiloidosis primaria. Relata clínica neurológica de cinco años de evolución consistente en: dolor y sensación de empastamiento en miembros inferiores, que evoluciona a torpeza motora para movimientos finos y pérdida de fuerza.
Se realiza estudio neurofisiológico: severa polineuropatía mixta de predominio en miembros inferiores de tipo desmielinizante y axonal. El estudio genético revelaba mutación del gen que codifica la síntesis de la TTR, una proteína plasmática sintetizada predominantemente en el hígado, siendo su transmisión autonómica dominante.
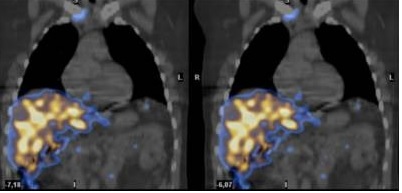
La ecocardiografía mostraba hipertrofia concéntrica del ventrículo izquierdo con una fracción de eyección del 60% y patrón diastólico restrictivo. En el estudio integridad celular miocárdica tras la administración de pirofosfato marcado, se aprecia un depósito intenso difusamente distribuido, sugestivo de afectación miocárdica global. La negatividad del estudio con Galio 67 y la positividad con pirofosfato inclina el diagnóstico hacia una amiloidosis hereditaria.
El estudio endoscópico macroscópico era de característica normales. Pero a nivel anatomopatológico de se evidenciaba un depósito amiloide en las biopsias rectales.
El resto de valoraciones (Otorrinolaringología, Neumología, etc.) fueron normales, por lo que, tras aconsejar la realización de estudio genético en pacientes de primer grado, el paciente se deriva al centro hospitalario de referencia para trasplante hepático.

Resultados
El único tratamiento eficaz para la PAF es el trasplante hepático (TH), tras el cual, la TTR anómala pasa a ser prácticamente indetectable en sangre, deteniéndose así la progresión de la enfermedad y observándose una lenta mejoría sintomática e incluso una progresiva disminución del depósito de amiloide.
La supervivencia postrasplante de estos pacientes ha demostrado ser similar a la obtenida en otros grupos de pacientes trasplantados, alcanzando más de un 70% al año y más de un 60% a los cinco años del TH.