
 Descargar número completo
Descargar número completo Download full issue
Download full issueCORRESPONDENCIA
Alberto Manuel García García
Hospital Universitario Virgen de la Victoria
29010 Málaga.
CITA ESTE TRABAJO
García García AM, Cobos Rodríguez J, García Ferreira A2, García Cortés M.Hepatotoxicidad aguda por paracetamol. RAPD Online. 2020;43(2):68-75.
Introducción
El daño hepático inducido por fármacos (drug-induced liver injury; DILI) es un efecto adverso frecuente dado que una gran cantidad de compuestos, incluyendo medicinas alternativas y productos de herboristería, son metabolizados a nivel hepático. Este daño puede manifestarse con mayor frecuencia como una hipertransaminasemia leve asintomática, sin embargo también puede inducir en algunos casos una enfermedad hepática grave que puede evolucionar a un fallo hepático fulminante con encefalopatía, coagulopatía e ictericia.
El acetaminofeno (N-acetil-p-aminofenol), también conocido como APAP en los Estados Unidos o paracetamol en Europa y el resto de países, se introdujo en la práctica clínica en 1955. Su empleo se ha extendido hasta el punto de convertirse en el analgésico/antipirético más utilizado en el mundo. Es más, es el componente de cientos de medicamentos de dispensación con y sin receta y la población suele subestimar su toxicidad. Además un porcentaje no desdeñable de pacientes ingiere cantidades excesivas de paracetamol porque no entienden la posología o no saben que se encuentra en más de uno de los medicamentos que están tomando.
Aunque a las dosis terapéuticas habituales (hasta 4000 mg al día) se considera un fármaco seguro[1],[2], desde 1966 se conoce que la sobredosificación puede producir necrosis hepática. Desde entonces la incidencia de hepatotoxicidad(HTX) por paracetamol ha ido en aumento y hoy día se trata de uno de los medicamentos que más frecuentemente causa DILI[1]-[4].
Epidemiología
En países como Estados Unidos o Reino Unido es el principal responsable de DILI siendo a su vez la causa más frecuente de fallo hepático agudo (con hasta el 50% de los casos) y suponiendo un 20% de las indicaciones de trasplante hepático[1]-[3],[5].
En España la intoxicación por paracetamol es mucho menos frecuente que en estos países aunque hay poco publicado al respecto. En un análisis retrospectivo a nivel nacional donde se analizó la causa de 267 casos de fallo hepático agudo, la sobredosis por paracetamol solo lo fue en 6 de ellos (2,24%)[6]. El fármaco más comúnmente responsable de DILI en España es la amoxicilina-ácido clavulánico.
Farmacocinética
El paracetamol está disponible en presentación tanto de liberación inmediata como de liberación sostenida. La dosis terapéutica máxima es de 80mg/kg/día en niños y 4g/día en adultos. Se absorbe rápida y completamente en el duodeno con una biodisponibilidad de entre el 60% y el 98%. La vida media de eliminación varía de dos a cuatro horas y se ha visto alargada en pacientes que desarrollan HTX. Se metaboliza principalmente en el hígado y tiene una distribución uniforme por todos los tejidos siendo eliminado en su mayor parte a través del riñón[7].
Toxicidad bioquímica
La toxicidad producida por el paracetamol es de tipo intrínseco o dependiente de dosis. El metabolismo de paracetamol tiene lugar en el interior de los microsomas hepáticos. A dosis terapéuticas, el 85-90% se metaboliza a conjugados de sulfato y glucurónido. Estos metabolitos conjugados se excretan posteriormente en la orina junto con aproximadamente un 2% que se excreta sin cambios[7]. El paracetamol restante se metaboliza mediante el sistema enzimático citocromo P450 hepático (en concreto por la subfamilia P2E1) por acción de las oxidasas mixtas generando un intermedio tóxico y altamente reactivo, el N-acetil-para-benzoquinoneimina (NAPQI)[8]-[12]. Las dosis adecuadas de paracetamol producen una pequeña cantidad de NAPQI que se conjuga rápidamente con glutatión hepático formando compuestos de cisteína y mercaptano no tóxicos que se excretan en la orina. Sin embargo, con dosis tóxicas las vías de sulfatación y glucuronidación se saturan y se desvía más paracetamol a las enzimas del citocromo y se metaboliza a NAPQI. Cuando las reservas de glutatión hepático se agotan en aproximadamente un 70-80%, el NAPQI comienza a formar enlaces covalentes con los grupos sulfhidrilo en las moléculas de cisteína y lisina formando aductos de proteínas que producen disfunción mitocondrial mediante un daño oxidativo que conduce a la necrosis hepatocelular[7],[10].
Factores que influyen en la HTX por paracetamol
Alcohol
La influencia que ejerce el alcohol como factor de riesgo de DILI por paracetamol es diferente en la ingesta aguda y en el consumo crónico de esta sustancia. Los autores de una guía de consenso americana[13] observaron que la toxicidad hepática por una sobredosis aguda de paracetamol fue menor en aquellos pacientes que realizaron la ingesta junto con alcohol en comparación con los que no lo consumieron: 5,1% frente al 15,2% respectivamente. Esto es debido a que el alcohol es un sustrato del citocromo P2E1 (CYP2E1) y tras una ingesta aguda compite por este con el paracetamol, confiriendo al paciente cierto papel protector frente al desarrollo de HTX.
En cambio, un consumo crónico de alcohol aumenta la síntesis y actividad del CYP2E1, siendo el efecto contrario al descrito y por lo tanto, aumentando el riesgo de toxicidad por el medicamento. Por otro lado, el consumidor crónico de alcohol está a menudo desnutrido, tiene más probabilidades de tener un período de ayuno reciente y suele tardar más tiempo en demandar atención médica, factores que pueden aumentar aún más el riesgo de daño hepático como se describe más adelante[14].[15].
Enfermedad hepática crónica
Los pacientes con una enfermedad hepática crónica que no ingieren alcohol de manera habitual parecen no tener un riesgo mayor de lesión hepática inducida por paracetamol[13],[16],[17]; sin embargo, el metabolismo de este se reduce en el hígado cirrótico. La vida media de eliminación en esta población de pacientes se prolonga un promedio de 2-2,5 horas y la actividad de la enzima citocromo P450 es más baja y no se puede inducir, lo que les confiere cierta hepatoprotección tras una sobredosis[18].
Medicamentos
El uso concomitante de ciertos medicamentos o productos de herboristería puede predisponer a la HTX en ausencia de sobredosis y puede empeorar el pronóstico de una sobredosis intencional.
Como ejemplos de medicamentos que alteran la actividad de CYP2E1 tenemos anticonvulsivantes como la Carbamazepina o la Fenitoína, antituberculosos como la Isoniacida o la Rifampicina y productos de herboristería como la hierba de san Juan o el Germander[3].
Por otro lado, fármacos como el Trimetoprim-Sulfametoxazol, los opioides y la Zidovudina pueden potenciar la HTX del paracetamol al competir por las vías de glucuronidación, lo que resulta en un aumento del metabolismo del paracetamol dependiente del CYP2E1[19].
A su vez, los opioides pueden reducir los niveles de glutatión y potenciar por tanto el daño hepático[7].
Otros
La desnutrición y un período de ayuno reciente predisponen al desarrollo de HTX dado que la glucuronidación (que depende normalmente de las reservas de carbohidratos hepáticos) se ve reducida y las reservas de glutatión están agotadas en estos pacientes[19].
La edad puede influir en el metabolismo del paracetamol. Los pacientes mayores de 40 años tienen más probabilidades de desarrollar DILI o insuficiencia hepática aguda (IGA), requerir un trasplante hepático o morir tras una sobredosis4. Sin embargo, los niños pequeños probablemente estén protegidos gracias a un mayor suministro y regeneración de glutatión y una mayor actividad de las enzimas de conjugación[20].
El humo del tabaco contiene inductores del CYP1A2, aumentando por tanto el metabolismo oxidativo. En una revisión danesa se observó que el consumo de tabaco es un factor de riesgo independiente para la mortalidad tras una sobredosis de paracetamol, sin importar la cantidad de tabaco consumido y siendo mayor aún si se asociaba a la toma de alcohol[21].
Varios estudios se han centrado en determinar la relación entre el daño hepático producido por el paracetamol y la presencia de obesidad o enfermedad hepática grasa no alcohólica (EHNA) dada la creciente incidencia de estas en los últimos años. Aunque ambas se asocian a una mayor actividad del citocromo P2E1, los estudios han mostrado un riesgo de 4 a 7 veces mayor de desarrollar DILI en pacientes con EHNA, no así en los pacientes con obesidad[22].
Manifestaciones clínicas
La clínica inicialmente es leve e inespecífica, no permitiendo predecir de forma fiable la HTX posterior. Sin embargo, los médicos debemos reconocer rápidamente la intoxicación por paracetamol para minimizar la morbi-mortalidad de los pacientes. El curso clínico de la sobredosis de paracetamol se puede dividir en cuatro etapas secuenciales (Tabla 1).
Tabla 1
Curso clínico de la sobredosis por paracetamol: Estadíos clínicos
Etapa I:En las primeras 24 horas después de una sobredosis, los pacientes pueden permanecer asintomáticos o presentar una serie de manifestaciones inespecíficas como náuseas, vómitos, diaforesis, palidez, letargo y malestar[19]. Las concentraciones séricas de AST y ALT suelen ser normales, pero pueden aumentar a las 8 a 12 horas después de la ingesta en pacientes con intoxicaciones graves[23].
Etapa II: El estadio 2 es aquel que transcurre desde las 24 a 72 horas tras la ingesta. Inicialmente los síntomas de la etapa I se resuelven y los pacientes parecen mejorar clínicamente, pero a medida que avanza esta etapa desarrollan hepatomegalia dolorosa[19].
De los pacientes que desarrollan lesión hepática, más de la mitad tendrá una elevación de la AST en las primeras 24 horas y el resto lo hará antes de 72 horas[23]. En esta fase también se pueden observar un alargamiento del tiempo de protrombina (TP) y aumento de la bilirrubina total junto con alteraciones de la función renal (aproximadamente en el 1-2% de los pacientes)[24].
Etapa III: Las anomalías de la función hepática alcanzan su nivel máximo a las 72 a 96 horas después de la sobredosis de paracetamol. Los síntomas sistémicos de la etapa I reaparecen junto con ictericia y encefalopatía hepática (EH). Se produce una marcada elevación de las enzimas hepáticas (a menudo mayor de 10.000 UI / L), un mayor alargamiento del TP y niveles de bilirrubina más elevados. También podemos observar hipoglucemia y acidosis láctica. La insuficiencia renal aguda se da en más del 50% de los pacientes con insuficiencia hepática severa. La muerte ocurre con mayor frecuencia en esta etapa, generalmente a partir de un fallo multiorgánico[19].
Etapa IV: Los pacientes que sobreviven a la etapa III inician generalmente al 4º día una fase de recuperación que se completa unos 7 días después de la sobredosis, aunque esta recuperación puede ser más lenta en los pacientes de mayor gravedad. Se produce una mejoría clínica y analítica progresiva, si bien la recuperación histológica puede tardar hasta 3 meses. Cuando se produce la recuperación, esta es completa; dado que no se ha descrito la enfermedad hepática crónica como secuela de la intoxicación por paracetamol[19].
En cuanto a la enfermedad renal aguda (Acute Kidney Injury;AKI), la incidencia de disfunción renal está relacionada con la gravedad del DILI.
Podemos observar elevaciones de urea y creatinina en sangre junto con proteinuria, hematuria y cilindros de células granulares y epiteliales en el análisis de orina. Se puede producir una necrosis tubular aguda o daño del endotelio vascular.
La función renal mejora hasta normalizarse entre 1 y 4 semanas tras la ingesta, aunque puede requerirse diálisis durante el episodio agudo. Parece que la administración de N-acetilcisteína, utilizada para disminuir la toxicidad hepática por paracetamol, no tiene efecto protector sobre el riñón[24].
Diagnóstico
Ante una sospecha de intoxicación aguda intencional o fortuita por paracetamol, lo primero que deberemos solicitar es una analítica básica que incluya hemograma, coagulación, bioquímica con transaminasas, gasometría y los niveles de paracetamol en sangre, ya que la concentración sérica del fármaco es la base para diagnosticar una intoxicación aguda y determina la necesidad o no de tratamiento. Se debe determinar a las 4 horas de la ingesta o a la llegada a urgencias si el tiempo desde la misma es mayor o se desconoce. En este último supuesto se deberá repetir 4 horas después.
Ante toda sospecha debemos obtener un historial que contenga el momento de la ingesta, la dosis, la intención (es decir, autolítica o no), el patrón de uso (dosis únicas o repetidas) y la existencia de comorbilidades que puedan predisponer al desarrollo de una lesión hepática (como el consumo crónico de alcohol, uso de anticonvulsivantes o ayuno reciente).
El riesgo de DILI se predice mejor al relacionar el tiempo de la ingesta con la concentración sérica de paracetamol. Sin embargo el historial de dosis no debe utilizarse para predecir la toxicidad, ya que los diferentes estudios no han encontrado correlación entre la cantidad de paracetamol ingerida y la concentración sérica del fármaco[25].
La relación entre los niveles y el tiempo desde la ingesta se evalúa de acuerdo con el nomograma de Rumack-Matthew modificado para determinar la necesidad de una terapia con N-acetilcisteína (NAC)[26] (Figura 1).
Figura 1
Nomograma de Rumack-Matthew. Rumack BH. Acetaminophen hepatotoxicity: the first 35 years. J Toxicol Clin Toxicol 2002; 40: 3. (Línea verde: toxicidad hepática posible; línea azul: toxicidad hepática probable; línea roja: alta toxicidad hepática; flecha naranja y franja violeta: ver texto).
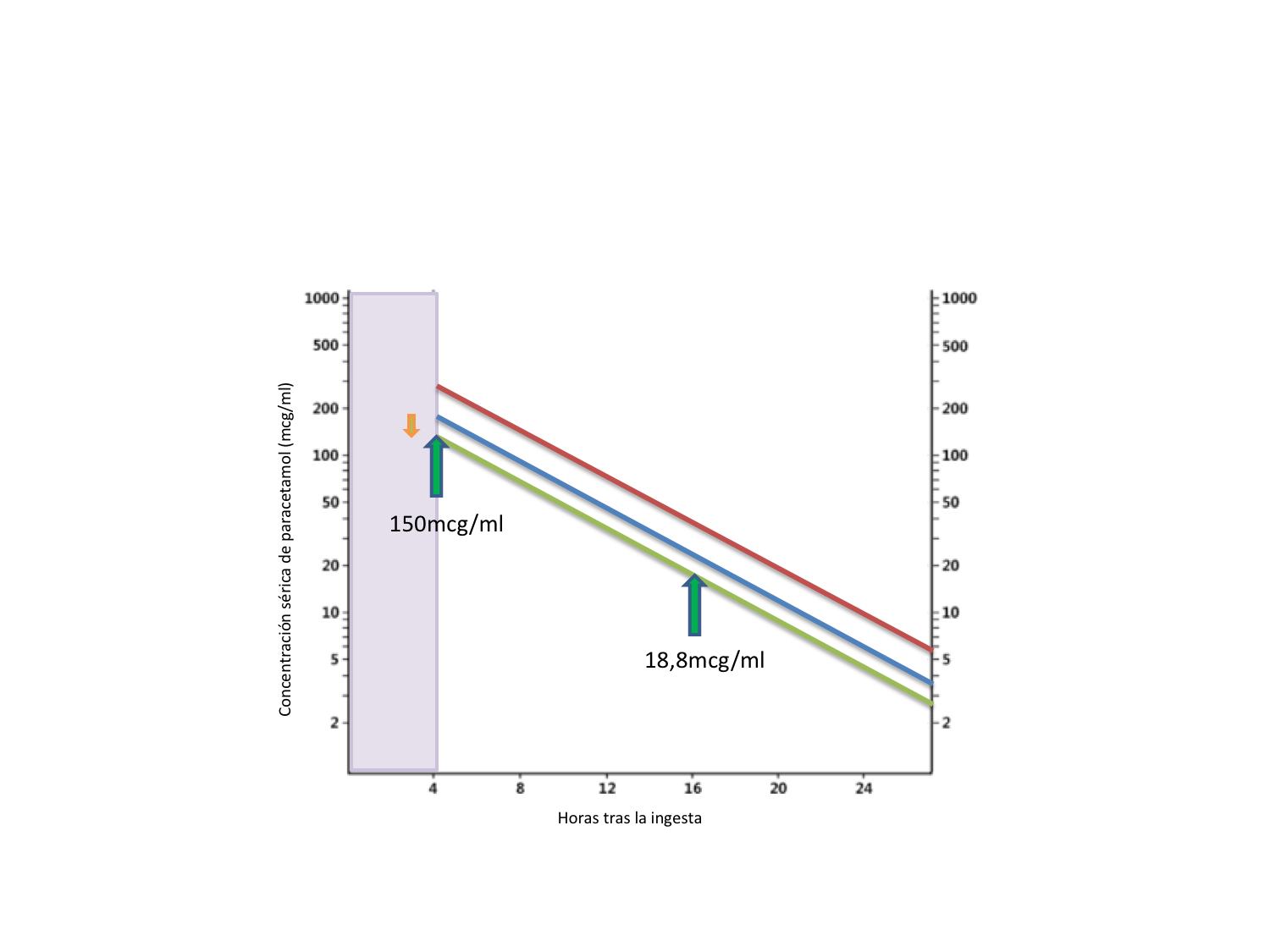
Las concentraciones séricas extraídas antes de las cuatro horas pueden no representar valores máximos y por tanto no deben utilizarse (franja violeta, figura 1). Usando este enfoque, los pacientes con concentraciones séricas de paracetamol por encima de la línea que conecta los 150 mcg/mL a las 4 horas y 18,8 mcg/mL a las 16 horas se consideran en "riesgo posible" de hepatotoxicidad (línea verde, figura 1) y tienen por tanto indicación de tratamiento con NAC[27]-[29].
El nomograma original, basado en un gran número de pacientes con sobredosis no tratados con antídoto, relacionó la concentración sérica de paracetamol con el momento de la ingesta como predictor de toxicidad. Los pacientes con concentraciones séricas por encima de la línea de toxicidad hepática probable (línea azul, figura 1) tienen un 60% de incidencia de DILI grave (definida como AST mayor de 1000 UI / L) y una tasa de mortalidad del 5%[30],[31].
Por último, los pacientes no tratados con concentraciones séricas por encima de la línea de alta toxicidad hepática (línea roja, figura 1) tienen una incidencia del 90% de toxicidad grave y una tasa de mortalidad de hasta el 24%[30],[31].
La línea de tratamiento en el nomograma se modificó posteriormente situándose un 25% más baja que la original[29],[32](flecha naranja, figura 1). Este margen de seguridad se creó para permitir variaciones en las mediciones de paracetamol entre los laboratorios y posibles errores en el tiempo estimado de la ingesta. La tasa de errores del nomograma con la línea modificada es extraordinariamente baja. El margen de seguridad del 25% probablemente protege a los pacientes susceptibles que tienen un mayor riesgo de desarrollar DILI (por ejemplo, usuarios crónicos de alcohol)[8],[33][34].
Nuevos biomarcadores
La detección de uno o más biomarcadores puede ser útil en los casos en los que el riesgo de toxicidad por paracetamol no esté claro. Se han detectado niveles elevados de aductos paracetamol-proteína mediante cromatografía líquida de alta presión en pacientes con sobredosis[35]. Por otro lado se han investigado diferentes marcadores de daño y muerte mitocondrial como la glutamato deshidrogenasa o el DNA nuclear o mitocondrial[11]. A nivel epigenético, los microRNA y en concreto el miRNA-122, se han identificado como potenciales biomarcadores precoces en el reconocimiento de la toxicidad inducida por el paracetamol en un estadio previo a la elevación de las aminotransferasas[36].
La detección de estos biomarcadores séricos puede ayudar en la toma de decisiones futuras de diseños terapéuticos y evaluaciones de trasplantes, así como para identificar futuras dianas terapéuticas.
Tratamiento
Tras una intoxicación aguda el paciente requiere una estabilización inicial y el tratamiento de los diferentes síntomas que pueden aparecer y que se han descrito previamente. Por otro lado, el tratamiento de la sobredosis de este fármaco se basa en la descontaminación gastrointestinal con carbón activado, en la administración de NAC que actúa como antídoto y en el tratamiento del daño hepático (Figura 2).
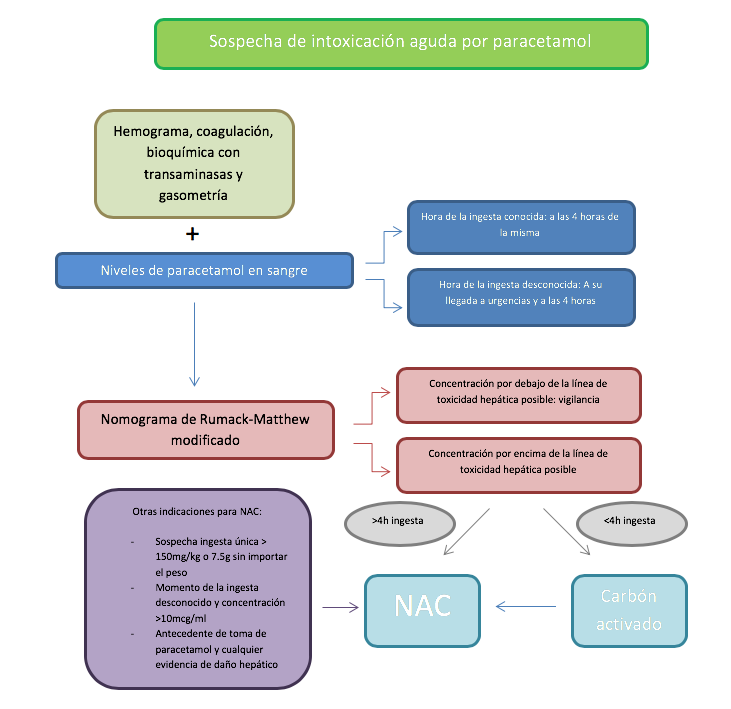
DESCONTAMINACIÓN GASTROINTESTINAL
Los pacientes adultos que se presenten menos de 4 horas tras una ingesta de paracetamol potencialmente tóxica probablemente se beneficien de la realización de una descontaminación gastrointestinal. En este sentido, está establecido el tratamiento con carbón activado a dosis de 1g/kg de peso hasta un máximo de 50 gr vía oral salvo que existan contraindicaciones[37].
Diferentes estudios han demostrado que el tratamiento con carbón activado reduce las concentraciones séricas de paracetamol, la probabilidad de desarrollar lesión hepática y de requerir tratamiento con NAC y que es más efectivo que el lavado gástrico o los vómitos inducidos[37],[38].
N-ACETILCISTEÍNA
Aunque persiste cierta controversia sobre su mecanismo de acción, la mayoría de los toxicólogos creen que la N-acetilcisteína previene la lesión hepática restaurando las reservas de glutatión hepático[39].
Las indicaciones de tratamiento con NAC son las siguientes:
Concentración sérica de paracetamol al menos 4 horas desde la ingesta por encima de la línea de tratamiento del nomograma modificado.
Sospecha de ingesta única de más de 150 mg/kg (7,5 g de dosis total sin importar el peso) si no se puede determinar la concentración sérica.
Momento de la ingesta desconocido y una concentración sérica> 10 mcg/ml.
Antecedentes de toma de paracetamol y cualquier evidencia de lesión hepática.
Si se administra NAC dentro de las 8 horas posteriores a la ingesta, la toxicidad grave es poco frecuente y la muerte es extremadamente rara. Incluso cuando se administra más tarde, en pacientes con evidencia de IHA, logra disminuir la mortalidad y mejora la función hepática y cerebral[29],[40],[41].
Las reacciones adversas más comúnmente asociadas a la administración de NAC son la anafilaxia con la administración intravenosa y los vómitos con la administración oral. Respecto a la anafilaxia, diferentes estudios prospectivos sugieren que entre el 10-20% de los pacientes tratados con NAC intravenosa desarrollan una reacción de hipersensibilidad no mediada por IgE. La gravedad de la misma puede variar desde eritema no pruriginoso hasta una insuficiencia respiratoria persistente[42]. En cuanto a la tolerancia digestiva, aproximadamente un tercio de los sujetos tratados con NAC oral desarrolla náuseas o vómitos. La tolerabilidad se puede mejorar mediante la dilución a una solución al 5% en coca-cola o zumo[43] o mediante la administración de antieméticos, donde se ha descrito que el Ondansetrón es un antiemético eficaz ampliamente usado en este contexto[44].
Si un paciente vomita en la primera hora posterior a una dosis oral, esta se debe administrar nuevamente. Los vómitos persistentes a pesar de la terapia antiemética son una indicación para la administración endovenosa.
Existen varios protocolos y vías de administración de NAC: (Tabla 2).
Tabla 2
Protocolos de tratamiento con NAC.
Protocolo de 20 horas IV: se ha utilizado en el Reino Unido desde la década de 1970. Es un régimen de dosificación complejo en el que se administra inicialmente una dosis de carga de 150 mg/kg IV durante 15 a 60 minutos. Tras esta dosis inicial se comienza con una perfusión a 12,5 mg/kg/h durante 4 horas que finalmente se reduce a la mitad durante 16 horas más[32],[45]-[47].
Protocolo simplificado de 20 horas IV: A diferencia del protocolo estándar, en el protocolo simplificado no se administra dosis de carga inicial si no que se comienza directamente con una perfusión a 50 mg/kg/h durante 4 horas que posteriormente se reduce a 6,25 mg/kg/h durante 16 horas más.
La experiencia en Europa, Australia y Estados Unidos y los resultados de un gran estudio retrospectivo indican que las reacciones anafilácticas durante el tratamiento con NAC IV pueden reducirse utilizando un régimen simplificado de dos bolsas en lugar del régimen tradicional de tres bolsas. En el estudio, las reacciones anafilácticas ocurrieron en el 10% de los 389 pacientes tratados con el régimen estándar, en comparación con el 4,3% de los 210 pacientes tratados con un régimen modificado[48].
Protocolo de 12 horas IV: Para el protocolo de 12 horas, la dosis de carga es de 50 mg/kg/h durante dos horas, seguida de una infusión a 20 mg/kg/h durante 10 horas más.
La perfusión se detiene una vez que el INR sea inferior a 1,3, la concentración de ALT inferior a 100 IU/L y la concentración sérica de paracetamol inferior a 20 mg/L. Si estos criterios no se cumplen, se debe continuar la perfusión de NAC.
Un pequeño ensayo controlado y aleatorizado comparó los pacientes tratados con el protocolo de 12 horas que tiene una tasa más lenta de infusión de dosis de carga con el protocolo estándar. Con el protocolo modificado se registró una tasa significativamente menor de vómitos y de reacciones anafilácticas[49].
Protocolo oral de 72 horas: el protocolo de dosificación oral de 72 horas se ha utilizado con éxito en los Estados Unidos durante más de 30 años. Consiste en administrar una dosis de carga de 140 mg/kg, seguida de repetidas dosis de 70 mg/kg cada cuatro horas, con un total de 18 dosis[32],[46],[47].
Duración del tratamiento tras una ingesta aguda:
Se deben determinar niveles de ALT cuando esté finalizando el protocolo. Si estos están elevados o la concentración sérica de paracetamol es detectable, se deberá continuar con una perfusión de 6,25 mg/kg/h IV o 70 mg/kg oral cada 4 horas y solicitar nuevos niveles de paracetamol y ALT a las 12 horas. Se puede detener el tratamiento cuando la concentración sérica de paracetamol sea indetectable, se compruebe un claro descenso de la ALT y el INR sea <2.
Trasplante hepático
La gran mayoría de los pacientes que desarrolla HTX tras una ingesta aguda de paracetamol se recupera con un tratamiento adecuado. En cambio, la admisión en un centro trasplantador es vital para aquellos pacientes que desarrollan datos de IHA como encefalopatía hepática (EH), coagulopatía o acidosis metabólica. Estos pacientes tienen una supervivencia libre de trasplante de tan solo el 36%[50].
Existen diferentes sistemas de puntuación para medir la severidad de la enfermedad hepática (KCH, MELD, SOFA, APACHE II), siendo el más utilizado el del King’s College. Los investigadores de este hospital desarrollaron un modelo pronóstico basado en dos grupos de pacientes con fallo hepático agudo, en unos inducido por el paracetamol y otros con una etiología diferente. En los pacientes con DILI por paracetamol la presencia de un pH menor de 7,3 o un lactato mayor de 3,5 a las 4 horas o de 3 a las 12 horas con una correcta reposición de volumen son criterios para indicar el trasplante. En ausencia de estos criterios, pacientes que presentan una EH III-IV, un INR >6.5 y una creatinina> 3.4 han mostrado tener peor pronóstico y por lo tanto serían así mismo candidatos a trasplante hepático.
Conclusiones
Una sobredosis por paracetamol puede ser fatal y la población general a menudo subestima los peligros potenciales del mismo. Las dosis máximas recomendadas diarias para evitar esta eventualidad son de 80/mg/kg en niños y 4g en adultos.
Existen una serie de factores de riesgo para desarrollar DILI tras una ingesta aguda de paracetamol, algunos de ellos son la ingesta crónica de alcohol, los medicamentos que afectan el sistema enzimático CYP2E1, la desnutrición y la edad avanzada.
El riesgo de toxicidad se predice mejor al relacionar el tiempo de la ingesta con la concentración sérica de paracetamol, el cual se evalúa de acuerdo con el nomograma de Rumack-Matthew modificado para determinar la necesidad de una terapia NAC que idealmente se deberá iniciar antes de 8 horas.
Las reacciones adversas más frecuentes tras la administración de NAC son las reacciones anafilácticas (IV) y los vómitos (VO) que son menos frecuentes con los protocolos simplificados.
El trasplante hepático es una opción terapéutica para aquellos pacientes que cumplan criterios de mal pronóstico.