
 Descargar número completo
Descargar número completo Download full issue
Download full issueCITA ESTE TRABAJO
Sánchez Moreno S, Diéguez Castillo C. Pancreatitis autoinmune tipo 1: un desafío diagnóstico con presentación inusual. RAPD 2025;48(2):88-90. DOI: 10.37352/2025483.6
Introducción
Presentamos el caso de un varón de 51 años que es diagnosticado de pancreatitis autoinmune tipo 1 a partir de criterios clínicos y serológicos con hallazgos radiológicos inusuales que entrañan una dificultad añadida al diagnóstico diferencial de la patología inflamatoria pancreática.
Caso clínico
Paciente varón de 51 años con antecedentes de diabetes mellitus tipo 2 y dislipemia que acude a Urgencias por cuadro de 48h de evolución de dolor en hipocondrio derecho irradiado a espalda e intolerancia oral, con pérdida ponderal asociada, sin ictericia ni fiebre. En el análisis de sangre a su llegada destaca una creatinina de 1.5 mg/dl, albúmina 3g/dL, hemoglobina 11 g/dL, PCR 12 mg/dL y leucocitosis con neutrofilia; con amilasa, bilirrubina total y resto del perfil hepático normal.
Se realiza un TC abdominal con contraste con hallazgos sugerentes de proceso inflamatorio en la encrucijada pancreatoduodenal, sin poder descartar etiología tumoral subyacente, con varias colecciones pancreáticas asociadas, la mayor de unos 5 cm, que produce obliteración de la primera porción duodenal y ectasia de la vía biliar extrahepática, con adenopatías locorregionales asociadas (Figura 1).
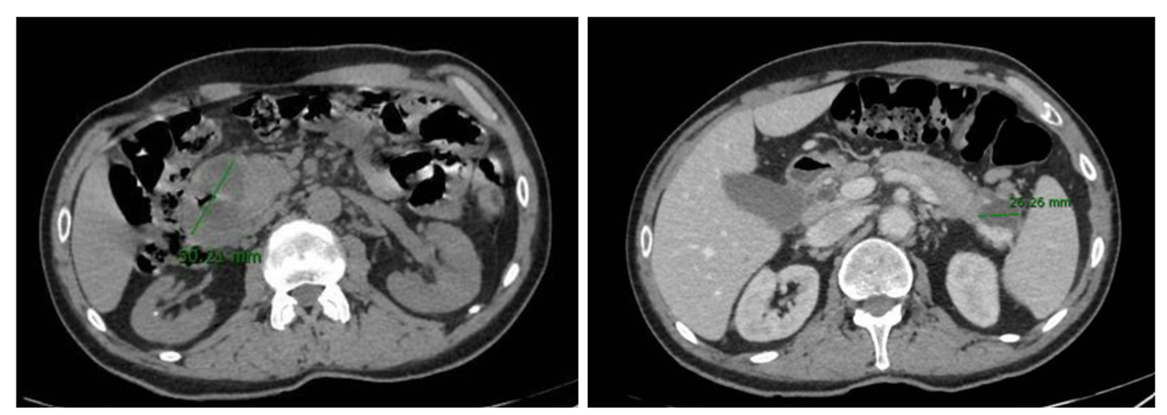
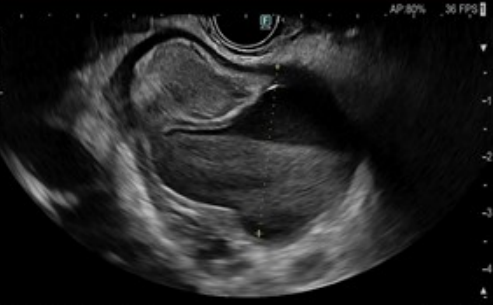
El marcador tumoral CA 19.9 resulta normal, y se realiza una ecoendoscopia (sin poder progresar a segunda porción duodenal por compresión extrínseca) que sugiere el origen quístico de las lesiones pancreáticas previamente descritas con un aumento homogéneo del páncreas sin otras lesiones focales, lo que orienta al origen inflamatorio del cuadro.
El análisis serológico detecta una inmunoglobulina G elevada (2090 mg/dL; VN 700-1600 mg/dL) con IgG4 de 1110 mg/d (>2 veces el VLSN). Posteriormente se completa el estudio con una RMN que objetiva un aumento difuso del páncreas con halo hipointenso periférico y colecciones de contenido líquido, sugiriendo estos hallazgos una pancreatitis de origen autoinmune dado el contexto clínico y analítico del paciente (Figuras 2 y 3) .
Figura 2
RMN abdominal. Disminución de tamaño de las colecciones pancreáticas tras inicio de tratamiento esteroideo.
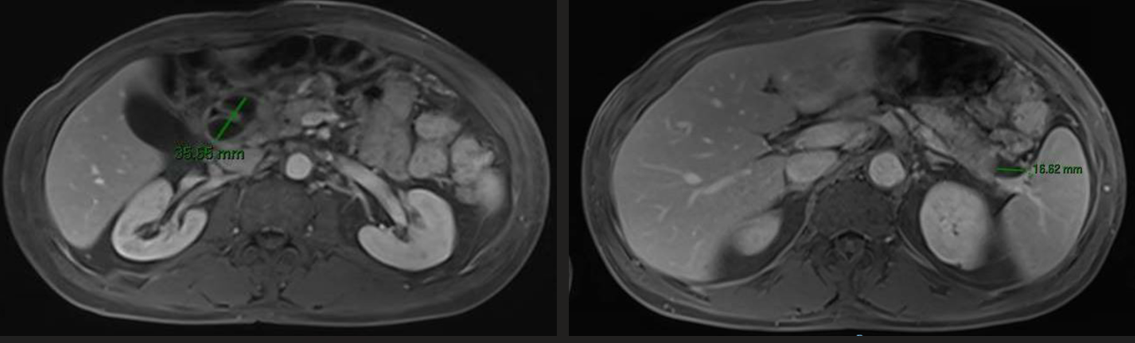
Se comienza tratamiento con corticoides presentando el paciente mejoría clínica con tolerancia oral progresiva y hallazgos radiológicos favorables a las dos semanas, con disminución de tamaño de las colecciones pancreáticas así como de las adenopatías locorregionales.
Discusión
La pancreatitis autoinmune (PAI) hace referencia a una enfermedad inflamatoria y fibrosante crónica del páncreas, de comportamiento benigno y de origen autoinmunitario, con respuesta a tratamiento con corticoides.
La PAI tipo 1 puede considerarse como una manifestación pancreática dentro del espectro de enfermedades por IgG4 y es frecuente la afectación de otros órganos extra pancreáticos, mientras que en la PAI tipo 2 no suele haber una elevación sérica de la IgG4 y puede relacionarse con la enfermedad inflamatoria intestinal[1].
Actualmente los criterios más empleados para hacer el diagnóstico de pancreatitis autoinmune son los del Consenso Internacional (2011), basados en la histología, hallazgos radiológicos, niveles séricos de IgG4[2], afectación de otros órganos extra pancreáticos y respuesta al tratamiento con corticoides. En función de su combinación se podrá establecer el diagnóstico probable o definitivo de PAI[3].
Mientras que la PAI tipo 1 puede ser diagnosticada con una alta precisión sin biopsia pancreática, para hacer el diagnóstico de la PAI tipo 2 casi siempre se necesita una confirmación histológica. En nuestro caso, la presencia de unos hallazgos radiológicos compatibles junto con la elevación significativa de los niveles de IgG4 y una respuesta favorable a corticoides, nos permitió realizar el diagnóstico de PAI tipo 1 con una alta precisión.
El diagnóstico diferencial principal de la PAI debe establecerse con el adenocarcinoma pancreático dado que las características clínicas de la enfermedad (síndrome constitucional, ictericia obstructiva, vómitos e intolerancia oral) y los hallazgos radiológicos (aumento de tamaño pancreático difuso o focal), a menudo plantean la sospecha de una neoplasia pancreática. De hecho, un porcentaje no desdeñable de pacientes sometidos a una duodenopancreatectomía por sospecha de cáncer son finalmente diagnosticados de PAI[1]. Es importante incluir también en el diangóstico diferencial de la PAI otras neoplasias que pueden afectar al páncreas como el tumor neuroendocrino y el linfoma pancreático.
Por otro lado, la presencia de colecciones agudas y pseudoquistes en la PAI se ha descrito clásicamente en la literatura como un hallazgo infrecuente que puede aportar confusión al diagnóstico; no obstante, en los últimos años se han publicado algunas series que señalan que la incidencia de estas lesiones en pacientes con PAI puede estar entre el 9.7% y el 22.4%[4],[5]. La aparición de lesiones quísticas pancreáticas en el seno de una pancreatitis autoinmune parece estar relacionada con la inflamación local de los segmentos pancreáticos afectados por la enfermedad junto con la estenosis de los conductos biliares intra pancreáticos que puede dar lugar a una retención secundaria de la secreción pancreática. Generalmente estas lesiones afectan al cuerpo y cola pancreáticos[5], pueden ser únicas o múltiples y, en muy raras ocasiones, de contenido hemorrágico o necrótico, pudiendo simular en ocasiones una neoplasia quística pancreática[6]. En nuestro caso, los niveles elevados de inmunoglobulina G fueron determinantes a la hora de orientar el diagnóstico.
La aparición de lesiones quísticas pancreáticas de manera sincrónica es una de las particularidades de nuestro caso, así como la compresión duodenal secundaria a la lesión de mayor tamaño (> 3cm) con repercusión clínica manifiesta en nuestro paciente, con imposibilidad para la ingesta oral y mejoría significativa tras el inicio de corticoides.
Con nuestro caso queremos resaltar la importancia de considerar la PAI en el diagnóstico diferencial del pseudoquiste y las colecciones agudas pancreáticas[7], y aportamos evidencia a favor del manejo precoz de estas lesiones en la PAI con tratamiento corticoideo dada la elevada tasa de regresión tras su inicio, siendo más excepcional el abordaje quirúrgico en estos casos.