
 Descargar número completo
Descargar número completo Download full issue
Download full issueCORRESPONDENCIA
Rosario del Pilar López-Segura
Unidad de Aparato Digestivo, Hospital U. San Cecilio. Avenida Dóctor Oloriz, nº 16
18012 Granada, España
TEL. 679105771
rosariopilarlopezsegura@hotmail.com
Sr. Director:
El dolor abdominal crónico puede ser un reto diagnóstico ante la ausencia de datos que orienten a su origen. Esto obligaría a pensar en otras entidades menos frecuentes y conocerlas para no someter a nuestros pacientes a una serie de pruebas complementarias, no exentas de riesgos, que podrían evitarse con la realización de un buen diagnóstico diferencial.
Presentamos el caso de una mujer de 37 años con crisis de dolor abdominal y líquido libre intraperitoneal de varios años de evolución, con múltiples ingresos hospitalarios e incluso laparotomía exploradora, que permanecía sin diagnóstico. El dolor era de localización centroabdominal y más frecuente en primavera. Había sido intervenida de apendicectomía sin hallazgos inflamatorios. Contaba episodios de hinchazón malar y enrojecimiento del dorso de ambas manos sin evidencia de traumatismo, que cedían espontáneamente. Como medicación habitual destacaba la toma de anticonceptivos orales (ACO). Tenía una hermana a la que le ocurría lo mismo y que también tomaba ACO, pero sin crisis abdominales.
El abdomen era depresible, doloroso en región centroabdominal sin signos de peritonismo.
- Analítica: PCR 2.10 mg/dl, leucocitos 22300/mm3 (91.1% neutrófilos).
- TAC toraco-abdómino-pélvico: Líquido libre intraperitoneal en cantidad moderada (Figura 1). Engrosamiento parietal de algunas asas de yeyuno, sin evidencia de dilatación intestinal (Figura 2).
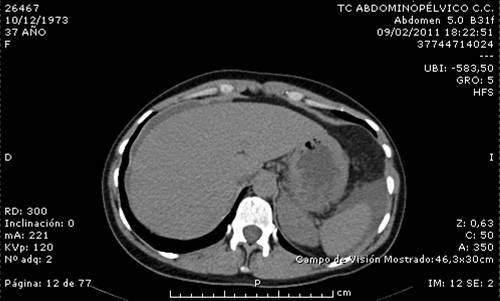
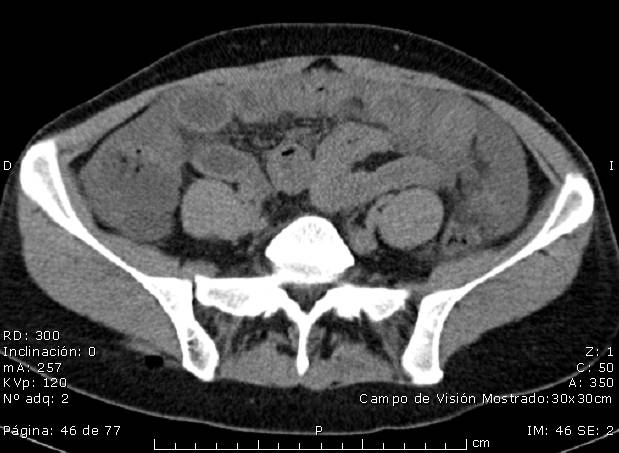
Ante los hallazgos clínicos se sospechó un angiodema hereditario, por lo que se solicitó Inhibidor C1 esterasa y complemento C3 y C4 que fueron negativos. Esto, unido a la toma de anticonceptivos orales, nos hizo sospechar un tipo III. Tras la retirada de los anticonceptivos orales la paciente se encuentra asintomática y en seguimiento.
El edema angioneurótico hereditario es una variante producida por mutación del gen C1 inhibidor (C1q) que favorece la inflamación y aparición de edemas recurrentes en piel y mucosas. Se transmite con herencia autosómica dominante[1]-[3]. Afecta a cara, laringe, aparato digestivo y extremidades[4], [5]. Puede confundirse con un abdomen agudo, y llevar a cirugía innecesaria. Existen 3 tipos de angioedema: Tipo I: El más frecuente (85%). La cantidad de C1q es inferior a la normal. Tipo II: niveles normales o elevados de C1q que no es funcionante. Tipo III: Descrito en el año 2000, aparece únicamente en mujeres y se asocia a la toma de anticonceptivos orales[6]. Presentan niveles normales de C1q que funciona bien, edemas en piel, episodios de dolor abdominal u obstrucción de vías aéreas altas. Las crisis agudas se tratan con C1 purificado o plasma fresco, y antifibrinolíticos. Reciben tratamiento crónico los pacientes que presentan uno o más episodios por mes (andrógenos o antifibrinolíticos), y deben evitar tomar IECA y ACO[7].
