
 Descargar número completo
Descargar número completo Download full issue
Download full issueCITA ESTE TRABAJO
Fernandez Carrasco M, Plaza Fernández A, Rodríguez Mateu A. Síndrome de Richter como manifestación de linfoma intestinal: una forma infrecuente de transformación agresiva de leucemia linfocítica crónica. RAPD 2026;49(2):64-66. DOI: 10.37352/2026491.1
Introducción
El SR representa una de las complicaciones más graves de la LLC y se caracteriza por la transformación a un linfoma agresivo, predominantemente DLBCL. Su incidencia anual se sitúa en torno a 0,5–1% y el riesgo acumulado a lo largo de la enfermedad se ha estimado entre 2–10%. La presentación habitual incluye crecimiento adenopático acelerado, síntomas B y elevación marcada de LDH, con supervivencias medias generalmente inferiores al año. La afectación del tracto gastrointestinal (TGI) es rara y puede dificultar el diagnóstico al imitar neoplasias primarias digestivas, enfermedad inflamatoria intestinal o procesos infecciosos[1]-[3].
Caso clínico
Varón de 54 años con antecedente de enfermedad de Hodgkin tratado con quimioterapia en 2003. En 2021 fue diagnosticado de LLC-B, inicialmente estadio 0. En 2022, ante duplicación linfocitaria, se detectaron deleciones 13q y 17p, iniciándose tratamiento con ibrutinib.
En enero de 2025 ingresó por dolor abdominal, astenia y pérdida de peso, objetivándose anemia severa, linfocitosis y esplenomegalia. En analítica destaca hemoglobina de 7,4 g/dL, LDH de 1.120 U/L, beta-2 microglobulina de 6,8 mg/L y calcio sérico corregido de 11,3 mg/dL.
El PET-TAC evidenció adenopatías hipermetabólicas, esplenomegalia y engrosamiento inespecífico a nivel de colon derecho y ciego. Se realizó colonoscopia, observándose válvula ileocecal engrosada y ulcerada con aspecto infiltrativo. La biopsia confirmó infiltración por neoplasia linfoide.
El estudio histológico fue compatible con SR por transformación de LLC a DLBCL, variante no centro germinal. La inmunohistoquímica mostró células B CD20+, CD5+, CD23+, BCL2+, MUM-1+, con índice proliferativo Ki-67 35–40%, sin evidencia de infección por virus Epstein–Barr. Se concluyó transformación agresiva con diseminación linfática y esplénica, con evolución fulminante y desenlace fatal.
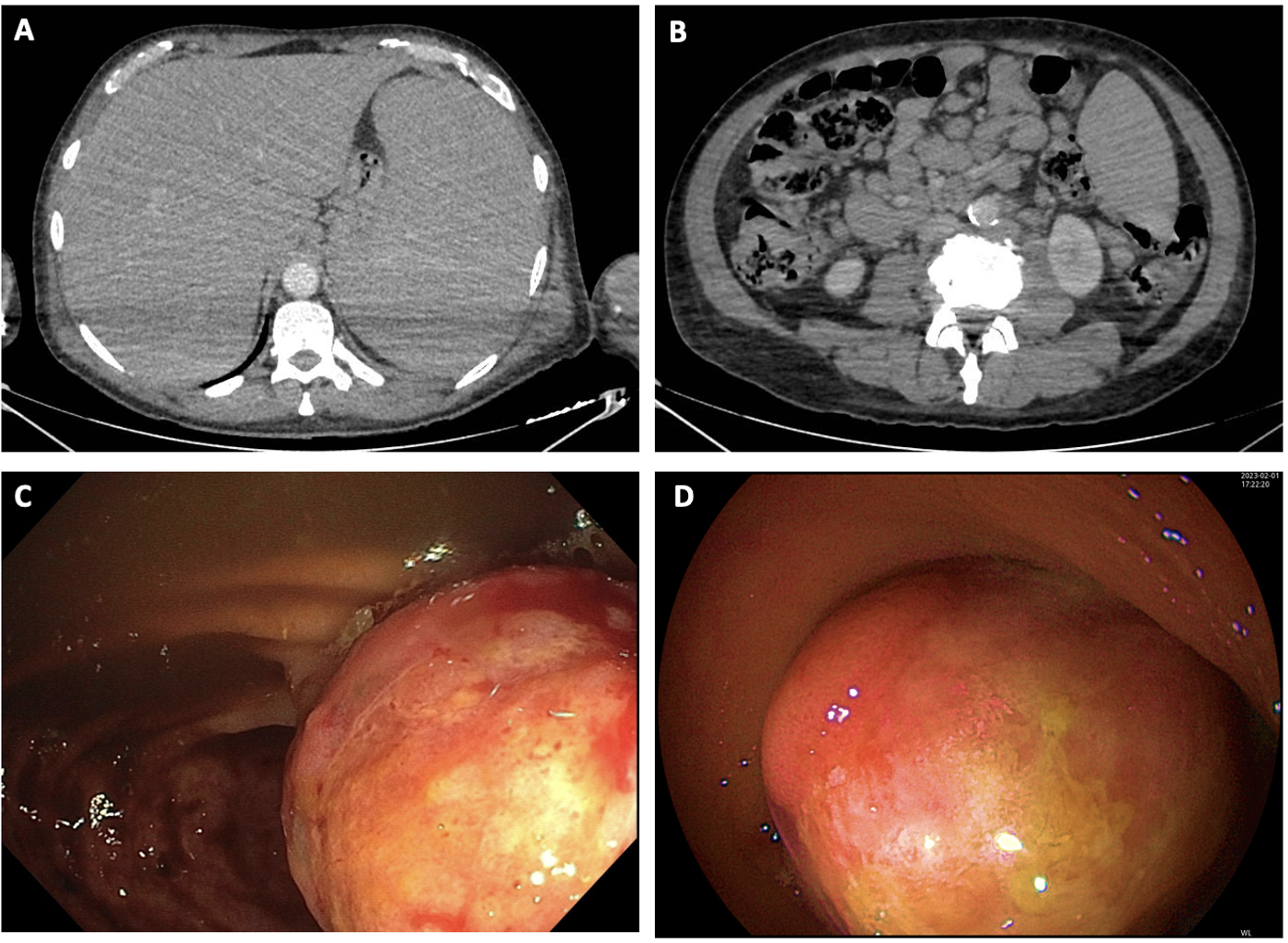
Figura 1
A) Imagen de TC abdominal que muestra hepatoesplenomegalia. B) Imagen de TC abdominal con presencia de adenopatía múltiples retroperitoneales y engrosamiento en colon derecho inespecífico. C y D) Imagen de colonoscopia con presencia de lesión neoformativa exofítica adyacente a válvula ileocecal con ulceración superficial.
Discusión
El SR es una entidad clínicamente heterogénea y de mal pronóstico. En torno al 90% de las transformaciones corresponden a DLBCL, mientras que la variante tipo linfoma de Hodgkin representa aproximadamente 5–10%. La agresividad biológica suele asociarse a alteraciones genéticas de alto riesgo, incluyendo TP53, NOTCH1 y MYC, implicadas en resistencia terapéutica y supervivencia inferior. La presencia de deleción 17p en nuestro paciente, ampliamente vinculada a disfunción de TP53, constituye un marcador pronóstico desfavorable y puede contribuir a la rápida progresión clínica[1],[2].
La afectación gastrointestinal por SR es excepcional y suele describirse en forma de lesiones infiltrativas o ulceradas en íleon terminal, válvula ileocecal o colon, con síntomas como dolor abdominal, anemia, pérdida ponderal o incluso sangrado digestivo. Su importancia clínica radica en que puede confundirse con patología digestiva primaria, demorando el diagnóstico. En estos casos, la combinación de síntomas sistémicos (síntomas B), datos analíticos sugerentes (LDH elevada, anemia, hipercalcemia ocasional) y hallazgos de imagen puede orientar la sospecha.
El PET-TAC resulta especialmente útil para localizar áreas de elevada actividad metabólica y guiar la obtención de biopsias del tejido más representativo. En nuestro caso, la correlación entre adenopatías hipermetabólicas y el engrosamiento colónico permitió orientar el estudio endoscópico y confirmar el diagnóstico[3].
El tratamiento del SR se basa en esquemas de quimioinmunoterapia tipo R-CHOP o R-EPOCH, con respuestas globalmente limitadas. En pacientes seleccionados se han evaluado combinaciones con venetoclax, inmunoterapia con bloqueo anti-PD1 y estrategias celulares como CAR-T, especialmente en enfermedad refractaria o de alto riesgo. Aun así, la supervivencia global continúa siendo pobre, reforzando la relevancia de la sospecha clínica temprana[4].
En el presente caso, la rápida evolución, el deterioro clínico y la agresividad de la enfermedad condicionaron una ventana terapéutica muy limitada, dificultando la instauración de tratamiento intensivo. Este hecho pone de relieve que la afectación gastrointestinal puede actuar como un marcador de agresividad y diseminación, y subraya la necesidad de actuar con rapidez diagnóstica ante manifestaciones digestivas atípicas en pacientes con LLC.
Conclusión
El SR es una complicación infrecuente pero altamente agresiva de la LLC. Aunque la afectación gastrointestinal es rara, debe considerarse ante síntomas abdominales asociados a clínica sistémica o hallazgos sugestivos en PET-TAC. La endoscopia con biopsia dirigida es esencial para confirmar el diagnóstico. Dado el pronóstico ominoso, especialmente en presencia de alteraciones de alto riesgo como deleción 17p/TP53, el reconocimiento precoz puede ser determinante para plantear estrategias terapéuticas antes de que se produzca deterioro clínico irreversible.